
Abstract
Clinicians and scientists at St George's University Hospital have collaborated to develop a classification algorithm for primary lymphatic anomalies. Instruction is offered on how to apply the algorithm in clinical practice to refine the diagnosis of primary lymphedema and guide on genetic testing and management. It can also be used to interpret mutation testing results of uncertain significance. The algorithm has evolved as more genes have been discovered, and it remains a “work in progress” as further discoveries are made. This transformational approach has revolutionized the understanding and classification of primary lymphatic anomalies.
Introduction
Primary lymphedema (PL) and lymphatic malformations (LMs) arise from faults in lymphatic development. In 1998 at St George's Hospital (SGH), London, United Kingdom, a clinic dedicated to PL was established. At that time phenotypes were not differentiated from one another except according to time of onset, that is, at birth, at puberty, and later onset. Any inherited PL with onset at birth would be called Milroy disease by default. As the only dedicated clinic in the United Kingdom for PL and with a referral network from throughout the United Kingdom, a research-based approach was introduced. Patients with similar clinical characteristics were grouped into cohorts and DNA was collected.
In 1995, the first gene shown to be involved in lymphatic development was FLT4 (VEGFR3) 1 creating the possibility of finding causal genes for human disease. PLs result from defects in genes involved in lymphatic vessel development. VEGFR3 was the first lymphedema gene to be identified in humans in 20002 after our publication of a gene for Primary Congenital Lymphedema mapping to the chromosome 5q35.3 region in 1999. 3
Through a process of rigorous phenotyping in the clinic, a second cohort of patients with PL was identified associated with distichiasis. Again, a locus was found by our group 4 with the gene, FOXC2, discovered by an American group. 5 Because the St George's (SG's) group provided essential linkage data for both discoveries, a series of research grants was obtained from the British Heart Foundation and this enabled a translational approach combining both basic and clinical research.
A Translational Approach
A clinical approach for the classification of PL was developed, based on phenotype. The SG's expertise and reputation from published research prompted increasing referrals of inherited lymphedema from throughout the United Kingdom. Patients were carefully categorized, DNA collected, and interrogated for gene mutations.
With careful phenotyping of patients by the clinical team (Professor Sahar Mansour, Dr Kristiana Gordon, and Professor Peter Mortimer) and gene analysis by Dr (now Professor) Pia Ostergaard and Professor Steve Jeffery, causal mutations were identified.
Knowing the genotype has enabled further investigation of the patients to define the full clinical characteristics of each type of PL as well as the mechanisms leading to the disease, for example, lymphatic valve failure.
Lymphedema Versus LMs
Interest in LMs inevitably followed as many of these birthmark anomalies can be associated with lymphedema but not always. LMs represent a structural abnormality of lymphatic vessels. Often the abnormality occurs in isolation with no communication with the main lymph conducting channels. In such circumstance swelling is due to the structural lymphatic vessel fault with the lymph trapped within it (atruncular lymphatic malformation). Conversely, if the malformation interferes with the main lymphatic channels then lymphedema can coexist because lymph is now trapped in the tissues, not just within the malformation (a truncular lymphatic malformation). 6
LMs are predominantly due to postzygotic mosaic mutations for which the phenotype can be variable but include lymphatic abnormalities. Somatic mutations are unlikely to be found in blood DNA. Genes such as PIK3CA and mutations within the Ras/mitogen-activated protein kinase (RAS/MAPKinase) pathway cause LMs, so a biopsy of the affected tissue is needed to provide a molecular diagnosis.
A Clinical Algorithm
It is so important to remember that PL is not just one disease, and there is much variation in the clinical manifestations. For many years the classification of PL was based on the age of presentation of the swelling with little consideration for associated clinical characteristics.
The SG's lymphedema team began developing a classification system for PL >15 years ago. Careful phenotyping, that is, looking at patterns of the swelling and other health problems led to the identification of five subgroups that shared the same broad category of PL.
These are as follows:
Lymphedema associated with other genetic syndromes, such as Noonan or Turner syndrome (where the lymphedema is not the overriding feature of the syndrome). Lymphedema with systemic, or internal, lymphatic problems. For example, pleural effusions, pericardial effusions, ascites, chylous reflux, protein losing enteropathy/intestinal lymphangiectasia or in utero swelling (fetal hydrops). Lymphedema that is congenital, so present at birth or within a few months of life (but no systemic involvement and the lymphedema is the predominant problem). Lymphedema that occurs later in life, after 1 year of age (but no systemic involvement and the lymphedema is the predominant problem). Lymphedema that may be associated with LMs, vascular malformations, or segmental overgrowth problems.
These five groups of PL are presented in the classification algorithm as color-coded sections, along with the individual subtypes, including the known genes. The algorithm was first published in 2010. 7
Over the years patients with similar phenotypes have been allocated to one of the five classification categories. Their DNA samples have been analyzed together to try and identify mutations common to the cohort, and this has proved very successful. Once a new gene has been discovered then close scrutiny and cross-checking of the clinical signs, natural history and inheritance patterns is performed to further refine an accurate phenotype for that genotype. This helps us in clinic to know what other health problems we need to screen for, for example, cardiac problems or leukemia, and also how best to manage the patient.
We consider the algorithm as a “work in progress” and it should be used as a living dynamic system that is constantly changing and being updated as new phenotypes and causal genes are identified. The SG's classification pathway was updated in 20138 and again in 2020. 9
The algorithm is designed to help clinicians categorize their patients and guide genetic testing, where possible. It involves “criteria matching,” that is, it uses specific findings for classification, and these are obtained through a process of taking a history, examination findings, and where possible incorporating results of investigation such as mutation testing and lymphoscintigraphy.
Making a Molecular Diagnosis
Making a molecular diagnosis is extremely helpful in the patient's management as it can help determine what other problems the patient is at risk of developing. Possible other health problems could include varicose veins, hydrocele, immunodeficiency, myelodysplasia or leukemia, congenital heart disease, scoliosis or spinal cysts, learning difficulties, eye abnormalities, renal abnormalities, and systemic lymphatic abnormalities (heart/lung/gut). Such pathologies are specific to the subtype. For example, a boy with Milroy disease might develop hydroceles in the future, but is definitely not at an increased risk of leukemia.
A molecular diagnosis will not only inform on inheritance patterns and likely prognosis but also guide the clinician on screening of diseases (as listed earlier) they are at risk of developing. The algorithm can help with this.
Knowing the gene fault can help determine mechanisms of disease. For example, a mutation in FOXC2 leads to lymphatic valve dysfunction and so reflux of lymph as the main cause for the lower limb lymphedema. It may also help with management. Understanding that reflux in both lymphatic vessels and veins is the mechanism with FOXC2 disease, explains why these patients respond well to intensive Decongestive Lymphedema Therapy and why swelling rebounds quickly afterward. Interestingly dealing surgically with the veins does not appear to help control swelling, indicating the dominant role of the lymphatics as the cause of edema.
Identifying genes can inform on their biological function and perhaps open up possibilities for targeted treatment of the lymphedema. Treatment for lymphedema remains physically based with surgery an alternative in some patients. Drug therapy is emerging as an option in select patients providing the gene is known. We can offer drug therapy targeting the PI3K/AKT/mammalian target of rapamycin signaling pathway (e.g. sirolimus) in patients with confirmed mutations in PIK3CA where disease appears to be still progressing. 10 In the RASopathies, for example, Noonan syndrome, caused by germline mutations in genes of the Ras-MAPK pathway, MEK-inhibitors offer promise to those with progressive lymphatic failure. 11
How Do You Use the Algorithm?
You start in the dark gray box, Primary Lymphatic Anomaly, and then move through the pathway working out which of the five subgroups your patient fits into depending on what problems they have (Fig. 1).
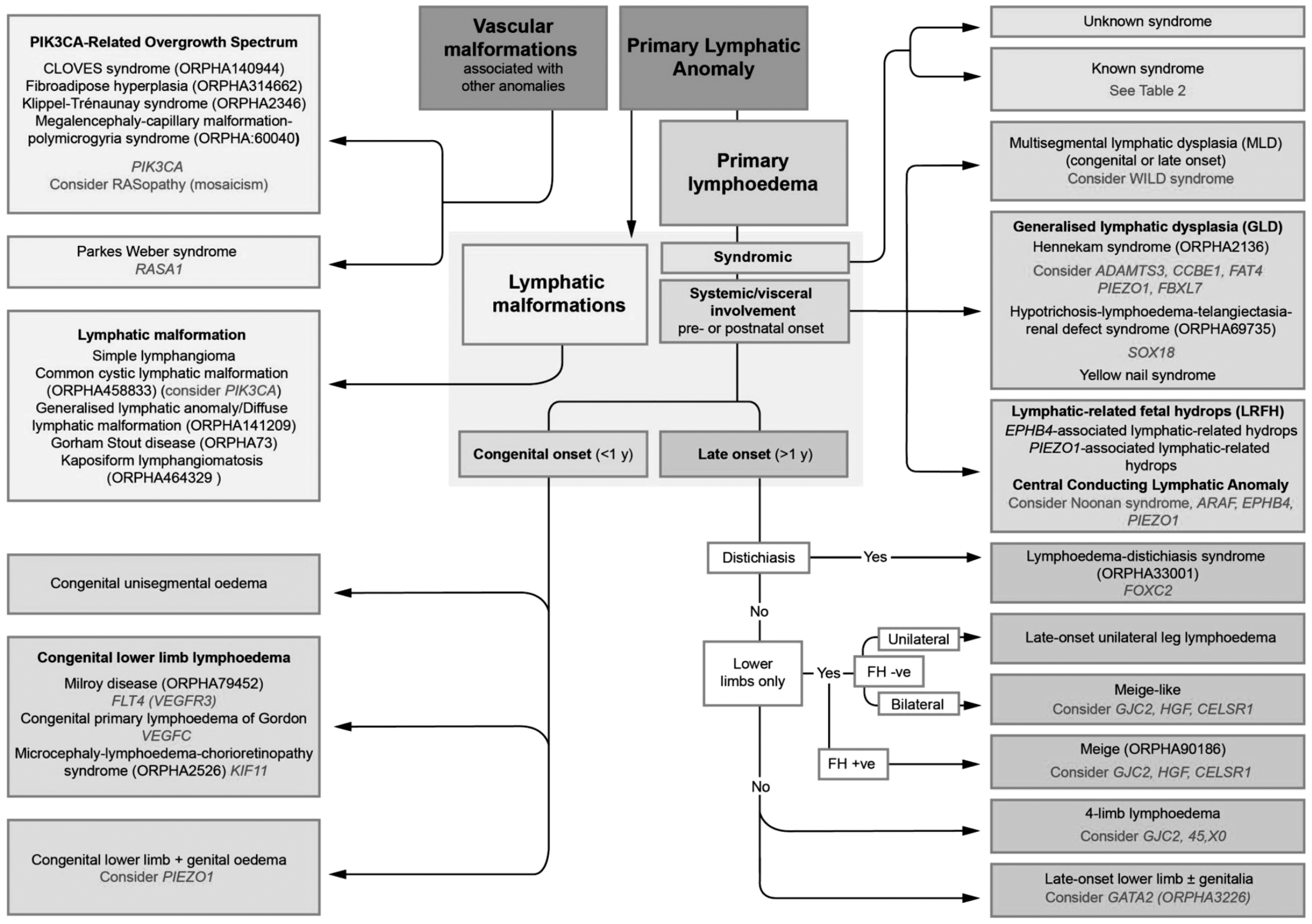
St George's classification algorithm for primary lymphatic anomalies (Image modified from “St George's Classification Algorithm” shared by St George's Lymphovascular Research Group under the CC BY-SA-4.0 International license).
Syndromic
The first question (blue box) is whether the patient fits into a known syndrome or appears to be “syndromic,” that is, a constellation of characteristics often including dysmorphic features. Testing for chromosomal abnormalities is usually worthwhile particularly if there is dysmorphism or learning difficulties.
Systemic involvement
If the patient does not have an underlying genetic syndrome, you move on to the pink section and consider if there are associated internal/systemic lymphatic problems such as pleural or pericardial effusions. Another pointer to the possibility of systemic involvement is the presence of fetal hydrops antenatally. Therefore, it is always important to enquire in the history if there was any extra fluid present in utero.
If you were referred a young patient with lower limb lymphedema and have excluded syndromes or systemic involvement from your history and examination, then you look at the green and purple sections. These mostly relate to lower limb lymphedema but do include genital and arm involvement too. The green section refers to congenital swelling, present at birth, whereas the purple sections classify swelling that comes on after the first year of life.
Congenital
Pedal lymphedema presenting at birth but with no “syndromic” or “systemic” features would make one consider Milroy disease and testing for mutations in VEGFR3 is worthwhile (green box). If a mutation is found in the VEGFR3 gene, then the diagnosis of Milroy disease is confirmed. The next step for the clinician is to use the information gathered to advise on natural history, prognosis and risks, and to guide management. Testing the parents will show whether the mutation in VEGFR3 has been inherited (10% of carriers are asymptomatic) or de novo (new in the child). If inherited, you can explain that future offspring and the patient's future children have a 50% chance of inheriting the condition, but can reassure the patient and their family that the swelling will remain confined to the lower limbs, although there may be a risk of varicose veins when older, and a third of affected males develop hydroceles.
Late onset
For late onset lymphedema (purple box) there are three main diagnoses to consider: lymphedema distichiasis syndrome (LDS), due to mutations in the FOXC2 gene, which may have varicose veins, congenital heart disease, cleft palate, spinal cysts, and renal problems; Emberger syndrome, due to mutations in the GATA2 gene, with widespread warts and monocytopenia/pancytopenia predisposing to myelodysplasia and acute myeloid leukemia; and Meige disease, which is the commonest subtype but no associated health problems for which no causal gene is yet known. One could ask why LDS and Emberger are not listed under syndromic? The answer is that the lymphedema is the dominant feature in both, whereas under syndromic the lymphedema is not considered a dominant feature.
The algorithm tells you to look for distichiasis first. If present, the diagnosis can only be LDS. Distichiasis refers to extra aberrant eyelashes arising from the inner eyelid. Even one aberrant eyelash is sufficient to consider this diagnosis. This abnormality is usually present at birth despite the lymphedema not manifesting until later in life usually late childhood/puberty but sometimes not until the fifth decade. This variability in age of onset of the swelling in LDS illustrates why distinguishing between the old terms of “praecox” and “tarda” forms of PL may not be useful. A diagnosis of LDS should prompt a search for any congenital heart or renal abnormalities.
A late onset, predominantly asymmetric, lower limb lymphedema with genital involvement should make one consider Emberger syndrome. Other characteristic features would include viral warts because of the underlying immunodeficiency. This phenotype illustrates why a blood test should be carried out. A low monocyte count would point to Emberger and the finding of a mutation in GATA2 confirms the diagnosis. A lifesaving bone marrow transplant should be considered to avoid leukemia. The family should be screened for the same GATA2 mutation before being considered as a bone marrow donor. It can literally be a lifesaving diagnosis to make, when death from leukemia can be prevented.
Lower limb lymphedema of late onset, particularly in a female, without any associated features suggest Meige lymphedema. Here, we could consider screening for HGF, GJC2, and CELSR1 although mutations have not been confirmed in a larger cohort of Meige cases.
Care must be taken to enquire about, and examine for, hand or upper limb lymphedema as it can be easily missed. Only by asking for swelling on the back of the hand, and not just fingers, will upper limb lymphedema be detected and, even then, four limb quantitative lymphoscintigraphy with quantification might be necessary for confirming the diagnosis. GJC2 mutations cause four limb lymphedema, with varicose veins but no other abnormalities and no systemic involvement.
Mutation Testing
Screening for a molecular diagnosis in all forms of PL might seem an easy option but, without careful phenotyping, the detection rate will be low.
We audited our PL clinic and of the 234 new patients seen in 1 year, we only found a causative mutation in 42% of the patients tested (25% overall including those not tested) (Fig. 2). We had not offered testing to a third of the patients because they had a phenotype of PL where the gene was not known, and we suspected the result would be negative (e.g., Meige disease).
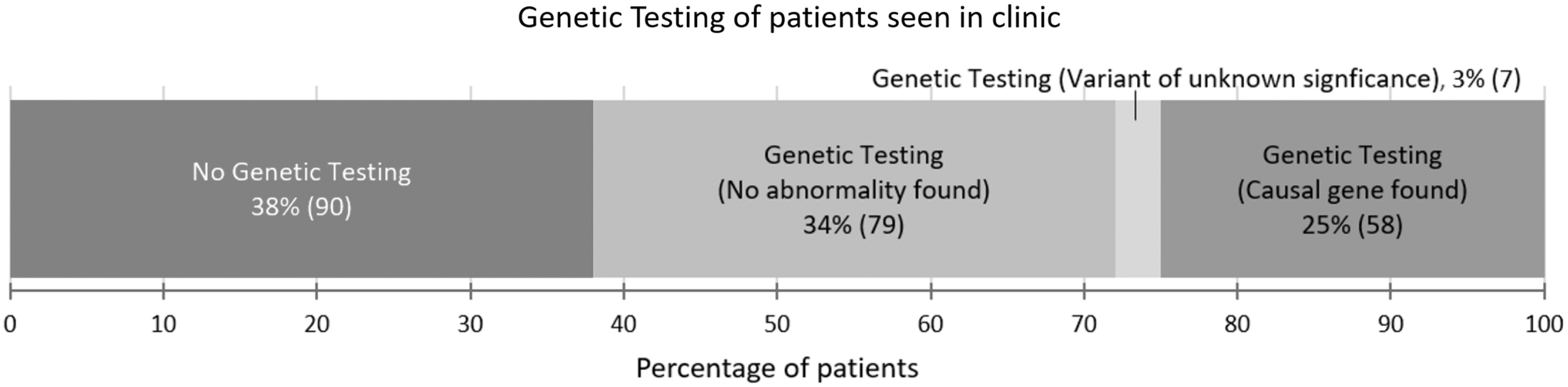
Audit of genetic testing results for 234 new patients seen in the primary lymphedema clinic for a 1-year period. A causal gene was only identified in 58 patients (25%).
These results have been replicated by other PL clinics worldwide, confirming the low pickup rate and indicating that there are many more causal genes to be discovered. We believe that matching a phenotype to a likely gene reduces wasteful and expensive testing.
Improvements in technology for genetic testing using next-generation sequencing have led to the introduction of testing for mistakes in several genes at once (the Lymphedema gene panel). However, this has also led to an increase in the detection of “variants of uncertain significance” (VUSs). We believe the algorithm helps enormously for the interpretation of these VUSs. Some centers are very quick to send off DNA for sequencing of all the patient's genes, but may struggle to interpret the results when the report says “there is a variant in Gene A, but we don't know if this is significant.” Correlation of where the patient fits into the algorithm will help the clinician decide if this gene variant or mistake is “real” or not. In other words, does the gene mistake match the clinical diagnosis. It is almost similar to using the algorithm in reverse, and so we believe it is a useful tool for all lymphedema clinics, even those that rely heavily on investigation results before examining the patient thoroughly first.
Impact of the Algorithm
This transformational approach pioneered at SG's has revolutionized the understanding and classification of primary lymphatic anomalies (i.e., PL and LMs). The combination of careful phenotyping and genotyping has enabled the evolution of the SG's classification pathway, which acts as an algorithm in the clinic to guide a specific diagnosis and management.
The impact of these developments has been to advise patients on what they can expect to happen from their disease and how best to avoid and manage the complications. The option of prenatal diagnosis and preimplantation genetic diagnosis is now possible.
The gene panel for PL, developed at SG's, is now available for use in the United Kingdom. Genomics England Limited (GEL) used information from the SG's lymphedema gene panel for their 100,000 genomes project for patients with PL.
The SG's classification pathway has been adopted by a number of lymphedema clinics in the United Kingdom and by the Primary and Paediatric Lymphedema Working Group (PPL-WG), part of the Vascular European Reference Network (VASCERN). The new Orphanet classification for primary lymphatic anomalies is based on the SG's classification and dovetails with the ISSVA classification for vascular anomalies.
Conclusion
The model of clinical care used by SG's has vindicated the benefit of combining basic with clinical science in a symbiotic relationship.
As more causal genes have been discovered so a molecular (genomic) diagnosis can be made more often. Knowing the gene permits knowledge of associated features, for example, venous or heart disease, informs on natural history of the disease, and enables understanding of gene function. Another benefit of a molecular diagnosis is that the genotype can be rigorously evaluated by investigation to refine the features of the phenotype.
Phenotyping using the algorithm improves the chances of finding yet more causal genes through research.
Investigation of the genotype can reveal likely mechanisms of disease. Understanding mechanisms provides the opportunity for targeting new therapies.
The SG's classification algorithm has evolved as more genes have been discovered. It remains “work in progress” and is designed to help clinicians phenotype their patients more accurately.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The research that led to the development of the SG's classification pathway was supported by the British Heart Foundation (BHF) [FS/06/063/21445; PG/10/58/28477; FS/08/006/25302; SP/13/5/30288], and a joint grant from the Medical Research Council (MRC) and the BHF [MR/P011543/1].