
Abstract
Background:
Generalized lymphatic anomaly (GLA), Gorham-Stout disease (GSD), kaposiform lymphangiomatosis (KLA), and central conducting lymphatic anomaly (CCLA) are rare, multisystem lymphatic disorders, referred to as complex lymphatic anomalies (CLAs). Their etiology remains poorly understood; however, somatic activating mutations have recently been discovered, and the results of targeted treatments are promising. This study aimed to elaborate on the phenotypic description of CLA.
Methods:
Thirty-six consecutive patients were recruited for the “GLA/GSD Registry” of the University Hospital of Freiburg, Germany (2015–2021). Clinical data were prospectively collected provided that a signed informed consent form was obtained. The latest proposed diagnostic guidelines were retrospectively applied.
Results:
Thirty-two patients (38% males) were included in the study; 15 GLA, 10 GSD, 3 KLA, and 4 CCLA patients were identified. Eighty-four percent already had symptoms by the age of 15 years. Osteolysis and periosseous soft-tissue infiltration were associated with GSD (p < 0.001 and p = 0.011, respectively), ascites and protein-losing enteropathy with CCLA (p = 0.007 and p = 0.004, respectively), and consumption coagulopathy with KLA (p = 0.006). No statistically significant differences were found in organ involvement, distribution of osteolytic lesions, number of affected bones and fractures. Twenty-five patients had complications; one patient with GLA died despite multimodal treatment. Spontaneous regression was seen in one patient with untreated KLA.
Conclusions:
CLA are rare, and their overlapping clinical presentations make differential diagnosis difficult. The characterization of our case series contributes to the phenotypic description and differentiation of these four clinical entities. A further understanding of their pathogenesis is crucial for evaluating targeted therapies and optimizing medical care.
Introduction
The term complex lymphatic anomalies (CLAs) has been introduced to describe a spectrum of rare, intractable lymphatic disorders with varying degrees of bone, soft-tissue, and organ involvement, unknown incidence, and poor prognosis. 1 According to the latest classification of the International Society for the Study of Vascular Anomalies, they are categorized as simple vascular malformations of the lymphatic system. 2 In the absence of a curative therapy, the treatment approach is multimodal. 3 The recent discovery of somatic activating mutations in the PIK3-AKT-mTOR and RAS-MAP-ERK signaling pathways in lymphatic malformations (LMs) has led to the use of mTOR inhibitors and MEK inhibitors, paving the way for precision medicine in the treatment of CLA. 4
Generalized lymphatic anomaly and Gorham-Stout disease
Formerly known as “lymphangiomatosis,” generalized lymphatic anomaly (GLA) is characterized by proliferation of dilated lymphatic vessels similar to common LM; however, GLA is a multifocal disorder that affects bones, soft tissue, and viscera. Nonprogressive osteolytic lesions are found in multiple noncontiguous bones of the axial and appendicular skeleton. 5 The mediastinum, spleen, liver, and lung are frequently affected; osteolysis usually affects the thoracic spine, humerus, femur, and ribs. 6
Gorham-Stout disease (GSD) is primarily an osseous disorder. The absence of visceral involvement was once a diagnostic criterion for GSD 7 ; however, soft tissue and viscera may be involved. The distribution of bone lesions in GSD is more regional, affecting fewer, contiguous bones, preferably of the axial skeleton. The cervical spine, base of the skull, mandible, clavicles, and ribs are the most commonly reported affected sites. 5
The diagnosis can be established both clinically and radiologically. 3 The key feature to radiologically differentiate GSD is severe progressive osteolysis with loss of the cortical bone. In fact, osteolysis in GSD can be very aggressive, sometimes leading to complete bone resorption (hence, the historic name “Vanishing Bone Disease”).5,8 Periosseous soft-tissue infiltration with intense enhancement upon administration of contrast on MRI is a marker of active disease in GSD. 9 In contrast, osteolytic lesions in GLA are well defined and restricted to the medulla, without active cortical bone destruction or periosseous soft-tissue involvement. 5 Pathological fractures (e.g., vertebral compression fractures) may complicate GLA, but they are more common in GSD. 9
Histologically, both conditions show proliferation of lymphatic channels in the medulla and cortex, with periosseous extension in GSD. 10 Bone destruction with active osteoclastic bone resorption characterizes GSD, while osteoclastic activity is rare in GLA. 5 A biopsy may be indicated if other osteolytic conditions (e.g., Langerhans cell histiocytosis, multiple myeloma, aneurysmal bone cysts, etc.) need to be excluded. A soft-tissue LM biopsy is recommended over a bone biopsy; a biopsy of the ribs, vertebrae, or base of the skull is contraindicated because of the high risk of complications (e.g., persistent pleural effusions, refractory lymphatic leakage, and cerebrospinal fluid leaks).1,3
Treatment for GLA and GSD is largely medical; both conditions respond to large extent to the mTOR inhibitors sirolimus and everolimus, sometimes with the addition of vincristine, while in cases of bony lesions the addition of bisphosphonates has shown a synergistic effect.11–14
Kaposiform lymphangiomatosis
Kaposiform lymphangiomatosis (KLA) is another multisystem LM with organ and bone involvement that exhibits a more aggressive clinical course, with significantly higher morbidity and fatality.15,16 KLA usually presents with respiratory symptoms caused by LM with extended pulmonary/mediastinal infiltration and effusions. Osteolytic lesions typically spare the cortex. 16 The defining clinical feature of KLA is consumption coagulopathy (i.e., Kasabach-Merritt-like phenomenon), similar to the Kasabach-Merritt phenomenon (KMP) observed in kaposiform hemangioendothelioma (KHE); a locally aggressive vascular tumor, with significant clinical and histological overlap with KLA. 3
While D-dimers may be moderately elevated, the risk of life-threatening bleeding is primarily determined by low fibrinogen levels and low platelet counts. Platelet transfusion is reserved for uncontrolled hemorrhage, as it may aggravate platelet trapping, with subsequent worsening of the coagulopathy. Cryoprecipitates may serve as an alternative option for patients with active bleeding or in preparation for surgical interventions.3,17–19 Therapeutic options for KLA include sirolimus,15,20 sometimes in combination with steroids or vincristine.21,22
As previously reported in GLA/GSD, the additional use of bisphosphonates for the treatment of osteolytic lesions can have a beneficial and/or synergistic effect in patients with KLA. 23 More recently, a somatic NRAS mutation was identified in KLA, with successful treatment with trametinib (MEK inhibitor).24,25
The assumption that KLA may be a subtype of GLA is not well established2,26; histologically, KLA is distinguished by the presence of intra-/perilymphatic abnormal, spindle-shaped lymphatic endothelial cells.16,27 Spindle cells tend to appear in sparse, poorly defined clusters in KLA, as opposed to the better defined vascularized nodules seen in KHE; both conditions may present at birth or in the first few years of life, but diffuse vascular lesions in the mediastinum and lungs, as well as refractory coagulopathy, are rather suggestive of KLA. 28 Serum levels of angiopoietin 2 are now used as a biomarker to distinguish KLA, as well as KHE with KMP, from GLA.29,30
Central conducting lymphatic anomaly
Central conducting lymphatic anomaly (CCLA) represents a wide spectrum of functional (e.g., dysmotility) or mechanical disorders (e.g., obstruction of the terminal portion of the thoracic duct) of the lymphatic vasculature, which cause inadequate lymph drainage, with subsequent stasis, lymphangiectasia, and reflux. 1 Clinical manifestations include chylous effusions, pulmonary lymphangiectasia, and protein-losing enteropathy (PLE). Osseous involvement is uncommon and demonstrates a channel-like appearance within the bone on MRI, mainly as a result of chylous reflux and dilated intraosseous channels. 9
Lymphangiographic findings (lymphangiectasia, lymphatic fluid reflux, and/or failure to empty into the thoracic duct or the subclavian vein at the thoracic duct outlet) are required to confirm the diagnosis. 3 In intranodal dynamic magnetic resonance lymphangiography (MRL), gadolinium is injected into the lymph nodes; the benefit over conventional lymphangiography is that MRL can also assess affected sites outside the lymphatic vessels (e.g., soft-tissue LM, organ involvement).3,31
In cases with an obstructive underlying mechanism, surgical reimplantation of the thoracic duct in an intrathoracic valved vein (venolymphatic anastomosis) is a treatment option. 32 Otherwise, CCLA treatment remains mainly supportive. Emerging evidence of an activating mutation in the RAS-MAP-ERK pathway implies that MEK inhibitors may be effective.33,34
Objectives
This study aimed to present a large case series of patients with CLA, primarily focusing on the phenotypic description. A secondary aim was to underline the diagnostic and therapeutic challenges, with an emphasis on the necessity for a multidisciplinary approach and a more thorough understanding of the underlying pathogenetic mechanisms of CLA.
Methods
Ethical approval and data collection
The GLA/GSD Registry of the University Hospital of Freiburg, Germany, was approved by the Ethics Committee of the University of Freiburg, Germany (310/14). Consecutive patients were recruited to the registry between 2015 and 2021, after signing a written informed consent (IC) form.
Data regarding clinical presentation, diagnostics, and treatment were prospectively collected in caregiver-completed case report forms. The following exclusion criteria were applied: revised diagnosis other than CLA, incomplete documentation even after retrospective review of the patients' charts, and absence of a signed IC form.
Diagnostic procedure
Clinical data were reviewed by two pediatric specialists (T.A.A. and J.R.), and radiological findings were independently reviewed by a pediatric radiologist (S.B.). The latest proposed diagnostic guidelines were retrospectively applied, 35 and patients were classified into the four clinical subtypes of CLA according to their clinical, radiological, and pathological findings. In case of diagnostic discrepancy, a final decision was reached by consensus with an effort to assign the diagnosis that best fit the clinical presentation.(T.A.A., J.R., F.K.).
Data analysis
Statistical analysis was performed using SPSS Statistics Edition 28. Descriptive statistical methods (median/range for quantitative variables; numbers/percentages for categorical variables) and nonparametric statistics were used. The Kruskal-Wallis test was used to compare the age at onset of symptoms, age at diagnosis, and time interval between symptom onset and diagnosis. GLA/KLA patients were compared with GSD patients in terms of the number of bones with osteolysis and the number of fractures, using the Mann–Whitney U test. For the comparison of categorical variables, the chi-square/Fisher's exact test was used. The level of statistical significance was set at 5% (p < 0.05), and all calculated p-values were two sided.
Results
Study sample description
A total number of 36 patients were recruited from 2015 to 2021; after reviewing all diagnoses, 2 patients with PIK3CA-related overgrowth syndrome and one patient with combined vascular malformation were identified and excluded from the study; a fourth patient was excluded due to missing data. In the final case series, we included 32 patients with CLA (age in years, median [range]: 20.9 [6 months–78.2]; 31% pediatric; 38% male).
Age at onset of symptoms did not vary significantly by diagnosis (age in years [range]: GLA [prenatal–46]; GSD [0–64]; KLA [prenatal–3]; CCLA [0–31], p = 0.123). In the vast majority of the patients (n = 27, 84%), symptoms had appeared by the age of 15 years; one-third of the sample (n = 10, 31%) already had symptoms at birth (Table 1).
Sample Characteristics
Medians (range) are provided, since the distribution was not normal for most of the groups (Shapiro–Wilk test, p < 0.05).
CCLA, central conducting lymphatic anomaly; GLA, generalized lymphatic anomaly; GSD, Gorham-Stout disease; KLA, kaposiform lymphangiomatosis.
Diagnosis
The diagnostic procedures included physical examination and laboratory testing, as well as extensive imaging studies to assess bone involvement (plain radiographs, CT, and MRI), soft-tissue LM or organ involvement (MRI with contrast), and effusions (ultrasound). Nine patients were also evaluated with MRL.
After retrospective application of the diagnostic guidelines proposed by Iacobas et al, 35 we identified 15 GLA, 10 GSD, 3 KLA, and 4 CCLA patients (Fig. 1). Patients with radiological evidence of cortical bone destruction were categorized as GSD; bone involvement in the remaining patients, if present, consisted of cortical thinning and osteolytic cystic lesions confined to the medulla (Fig. 2).
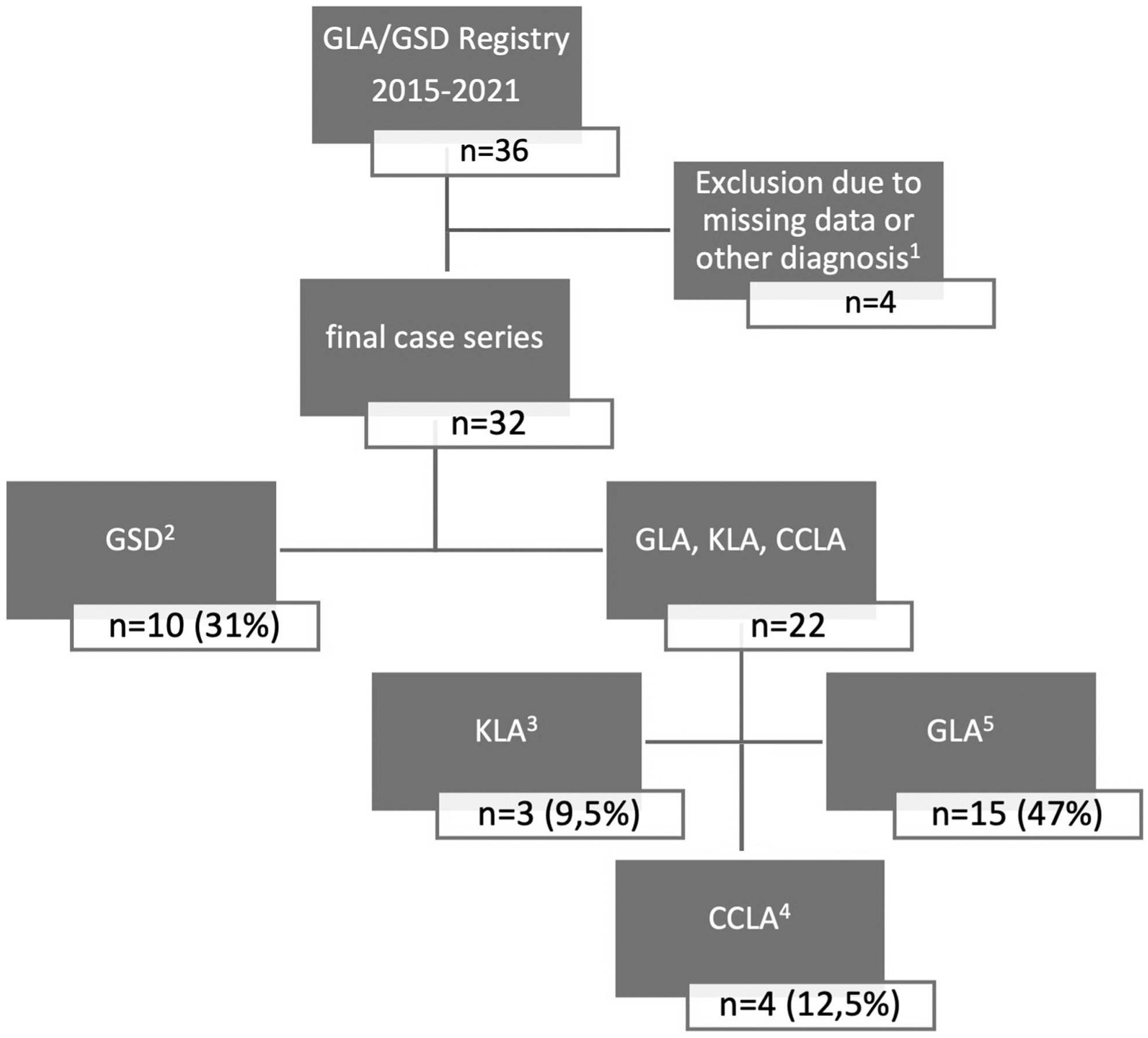
Flowchart of the diagnostic procedure of the case series. 1 Reasons for exclusion: insufficient documentation n = 1, other diagnosis n = 3 (combined vascular malformation n = 1, PIK3CA-related overgrowth spectrum n = 2). 2 All patients with radiological evidence of cortical bone destruction were identified and were assigned a GSD diagnosis. 3 A KLA diagnosis was histologically established in one patient, and two more patients were clinically diagnosed. 4 Four patients were clinically diagnosed with CCLA, exhibiting a constellation of PLE (n = 3), chylous effusions (chylous ascites n = 4, chylothorax n = 3), lymphedema (n = 3), and absence of bone involvement (n = 4), with typical MRL findings confirming CCLA diagnosis. 5 15 patients having a typical combination of clinical manifestations were diagnosed with GLA. CCLA, central conducting lymphatic anomaly; GSD, Gorham-Stout disease; GLA, generalized lymphatic anomaly; KLA, kaposiform lymphangiomatosis; MRL, intranodal dynamic magnetic resonance lymphangiography; PLE, protein-losing enteropathy.

Osseous involvement in GLA and GSD.
GLA was diagnosed in patients with a combination of typical clinical and radiological features. A KLA diagnosis was assigned to patients with characteristic clinical or histological findings. Patients with a constellation of manifestations indicative of CCLA and a lymphangiographic confirmation of the diagnosis were categorized as CCLA (Table 2).
Overview of the Entire Case Series
These four patients (2 GLA, 1 GSD, and 1 KLA) exhibited MRL findings consistent with CCLA; however, their clinical manifestations were typical of a different diagnosis, and were thus categorized accordingly.
A KRAS mutation was identified in a patient clinically diagnosed with GLA, who was unresponsive to mTOR inhibitors, and responded well to treatment with a MEK inhibitor. KRAS mutations have been reported in CCLA, but not in GLA. However, a CCLA diagnosis could not be confirmed on MRL, and the patient was categorized as GLA; the presence of cystic lesions in the thorax, abdomen, liver, and spleen supported this diagnosis.
Two patients were clinically diagnosed with KLA: patient 9 due to consumption coagulopathy and almost complete infiltration of both lungs, and patient 32 due to LM and consumption coagulopathy at birth. One more patient was histologically diagnosed with KLA (patient 26).
This patient had histological findings of KLA with spindle cells; however, a GSD diagnosis was more representative of the patient's clinical findings.
GI, gastrointestinal; LM, lymphatic malformation; m/f, male/female; MRL, intranodal dynamic magnetic resonance lymphangiography; PLE, protein-losing enteropathy.
Age at diagnosis was not significantly different among the four CLAs (p = 0.083). Patients with GLA tended to remain longer undiagnosed after the onset of symptoms (years until diagnosis, median [range]: GLA 2.3 [0–39] versus GSD 0 [0–7]; KLA 0.5 [0–2]; CCLA 0 [0–1.5]), but not at a statistically significant level (p = 0.253). The male-to-female ratio was even (1:1.66) among the four diagnoses.
A biopsy was performed in 18/32 patients (56%); histopathology demonstrated anastomosing endothelium-lined spaces that stained positive for the lymphatic markers D2-40 and Prox1, consistent with lymphatic vessels. Genetic testing of available tissue was performed in 11/32 patients (34%). One patient with GSD had a germline mutation in TSC2, in the context of a personal and family history of tuberous sclerosis.
Two activating somatic mutations in the RAS signaling pathway were identified in a patient who was clinically diagnosed with KLA and a patient with GLA without osteolysis. Since the number of patients with genetic testing is small, the main focus of this study remains the phenotypic description of CLA. Finally, none of our patients had a family history of vascular malformations.
Clinical manifestations
Thoracic or mediastinal LM were the most common clinical manifestation overall (n = 20, 63%). The most commonly affected organ was the spleen (n = 17, 53%), in the form of splenomegaly and/or multiple cystic lesions, followed by the lungs (n = 13, 41%) and gastrointestinal tract (n = 6, 19%). Organ involvement was similarly distributed in the four diagnoses (p = 0.508).
Four patients (GLA, n = 2; KLA, n = 2) exhibited prenatal onset of disease; two patients (GLA, KLA) had sonographic evidence of vascular malformation and two patients (GLA, KLA) presented with hydrops fetalis (1 GLA, 1 KLA).
Nineteen (59%) patients had osteolytic lesions (Fig. 2). Osseous involvement and periosseous soft-tissue infiltration were associated with GSD (p < 0.001 and p = 0.011, respectively). The number of affected bones did not differ significantly between the GSD and GLA/KLA groups (p = 0.774). The most commonly affected sites were the spine, pelvis, and lower limbs, with no statistically significant differences between the two groups. In the majority of cases with bone involvement (15/19, 79%), both the axial and appendicular skeleton were involved.
Lymphedema was present in 12/32 patients (38%) and 3 of 4 patients with CCLA (p = 0.482). Effusions were common in our sample (n = 19, 59%; pleural effusion: n = 17, 53%; ascites: n = 10, 31%). The presence of effusions did not vary significantly among the four diagnoses (p = 0.314), except for ascites, which was associated with CCLA (p = 0.007). Pericardial effusion was only observed with concomitant pleural effusion, and ascites was more common in patients with pleural effusion, but neither of these associations reached statistical significance (p = 0.229 and p = 0.06, respectively) (Tables 2 and 3).
Clinical Manifestations and Complications of Complex Lymphatic Anomalies
A case of meningitis was documented in a patient with GSD and osteolytic lesions in the base of the skull.
Treatment, follow-up, and complications
The majority of the patients (n = 24, 75%) required multimodal treatment, including interventional procedures (n = 17, 53%), medication (n = 21, 66%), and supportive measures (n = 18, 56%). The remaining patients were either operated once (n = 4, 12.5%: surgery at the site of LM in 3 GLA patients; decompressive surgery due to cervical spine lesions in 1 GSD patient) or clinically followed up without receiving treatment (n = 4, 12.5%: 2 GLA and 2 KLA patients).
Interventional procedures included: operation at the site of the LM, n = 14; sclerotherapy, n = 2; laser, n = 4; orthopedic surgeries, n = 6; management of effusions, n = 14. In addition, one patient with obstructive CCLA underwent venolymphatic anastomosis (Fig. 3).
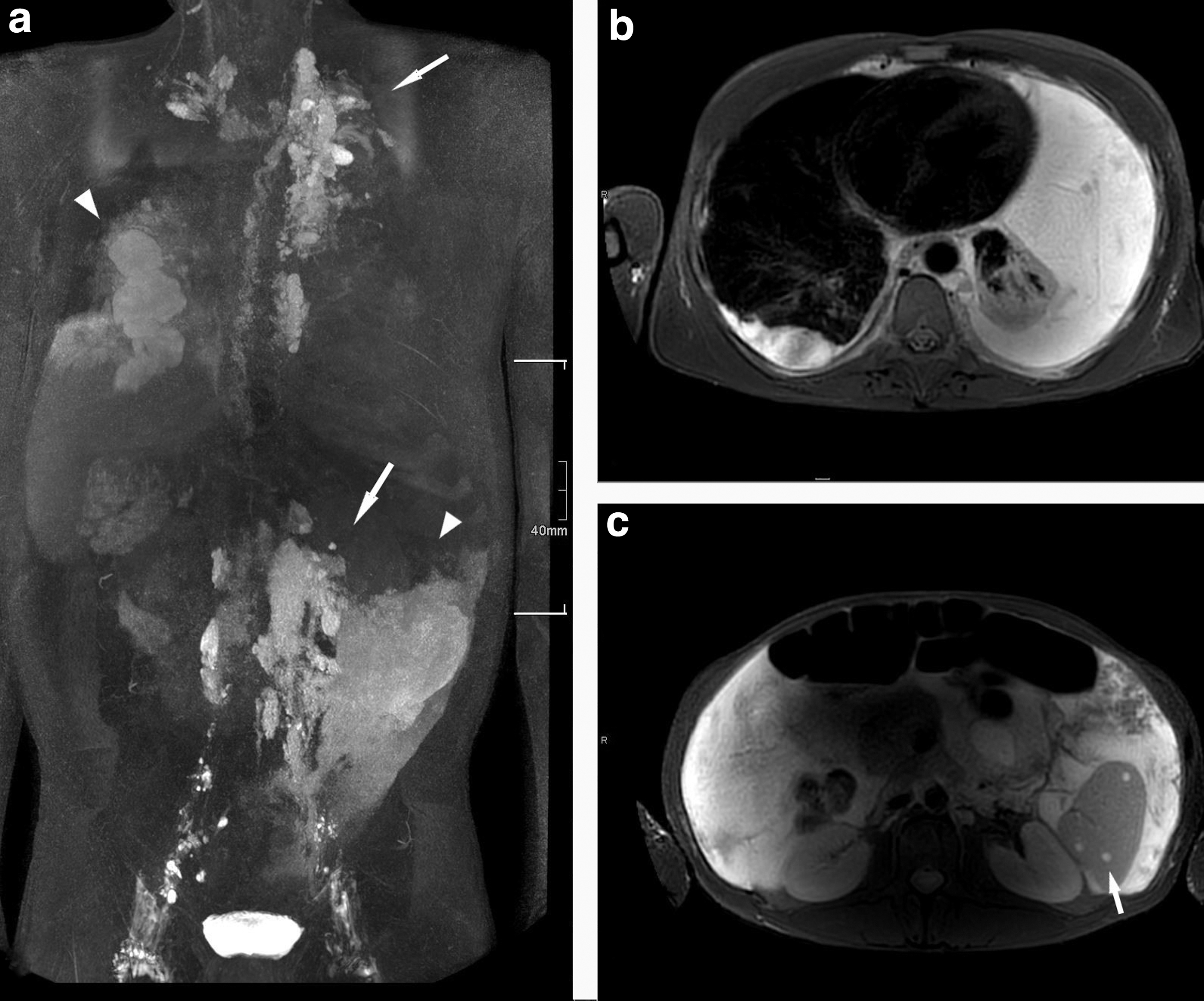
Radiological findings of a 35-year-old patient with CCLA (see Table 2, patient 7).
Medications included propranolol, corticosteroids, interferon-alpha 2α, vincristine (combined with methotrexate or 6-mercaptopurine), octreotide, and bisphosphonates; 17 patients (53%) received off-label individualized treatment (mTOR inhibitors, n = 16; MEK inhibitors, n = 1).
Supportive measures included compression therapy and/or lymph drainage in patients with soft-tissue LM and/or lymphedema (n = 9), and nutritional measures (low-fat medium-chain triglyceride diet, n = 13; human albumin substitution and/or parenteral nutrition due to PLE, n = 3).
Treatment response was documented for 22 of 28 treated patients; despite multimodal treatment, only 4 patients had clinical/radiological signs of improvement, 17 patients stabilized, and 1 patient with CCLA deteriorated under treatment. Interestingly, a spontaneous regression was observed in one patient with untreated KLA (Fig. 4).
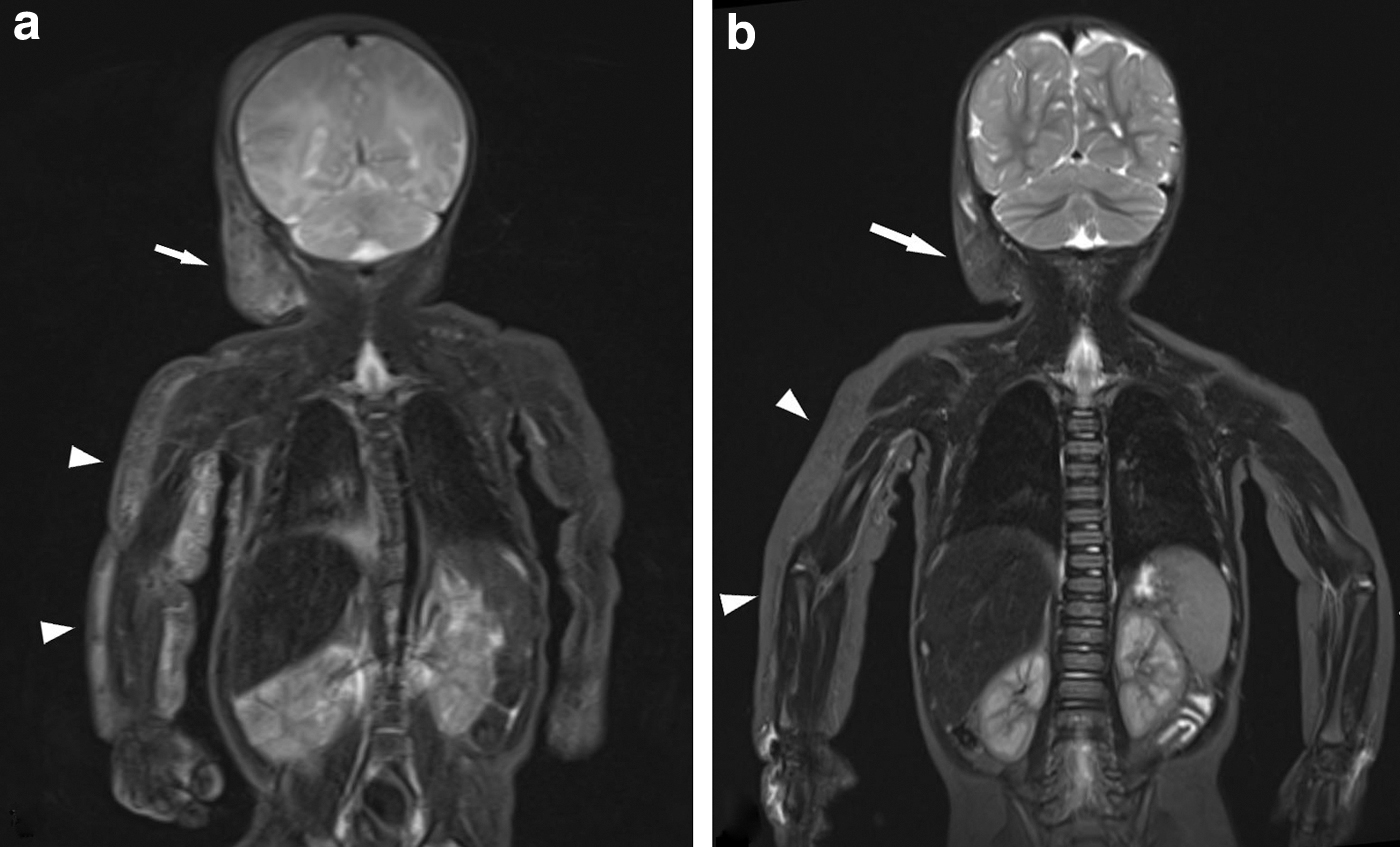
Infant with KLA and spontaneous regression (see Table 2, patient 32).
The majority of our patients (n = 25, 78%) had at least one complication (Table 3). The most common complication was severe infection (n = 14, 44%).
Progressivity of bone lesions was more common in GSD than in GLA/KLA (p = 0.170). Almost one-third of patients with osteolytic lesions of the spine developed scoliosis (5/16, 31%; p = 0.530). Bone fractures occurred in half of the patients with osseous involvement (9/19, 47%); neither the incidence nor the absolute count of bone fractures differed significantly between the GSD and GLA/KLA groups (p = 0.370 and p = 0.212, respectively).
Respiratory failure (n = 6, 19%) was associated with the presence of pleural effusion (p = 0.019), while no association with diagnosis, lung involvement, or presence of LM in the thorax/mediastinum was found (p = 0.355, p = 0.194, and p = 0.37, respectively).
PLE was associated with CCLA (p = 0.004) and consumption coagulopathy with KLA (p = 0.006).
One patient with GLA and thoracic involvement died of respiratory complications during the time period of the registry (Fig. 5).
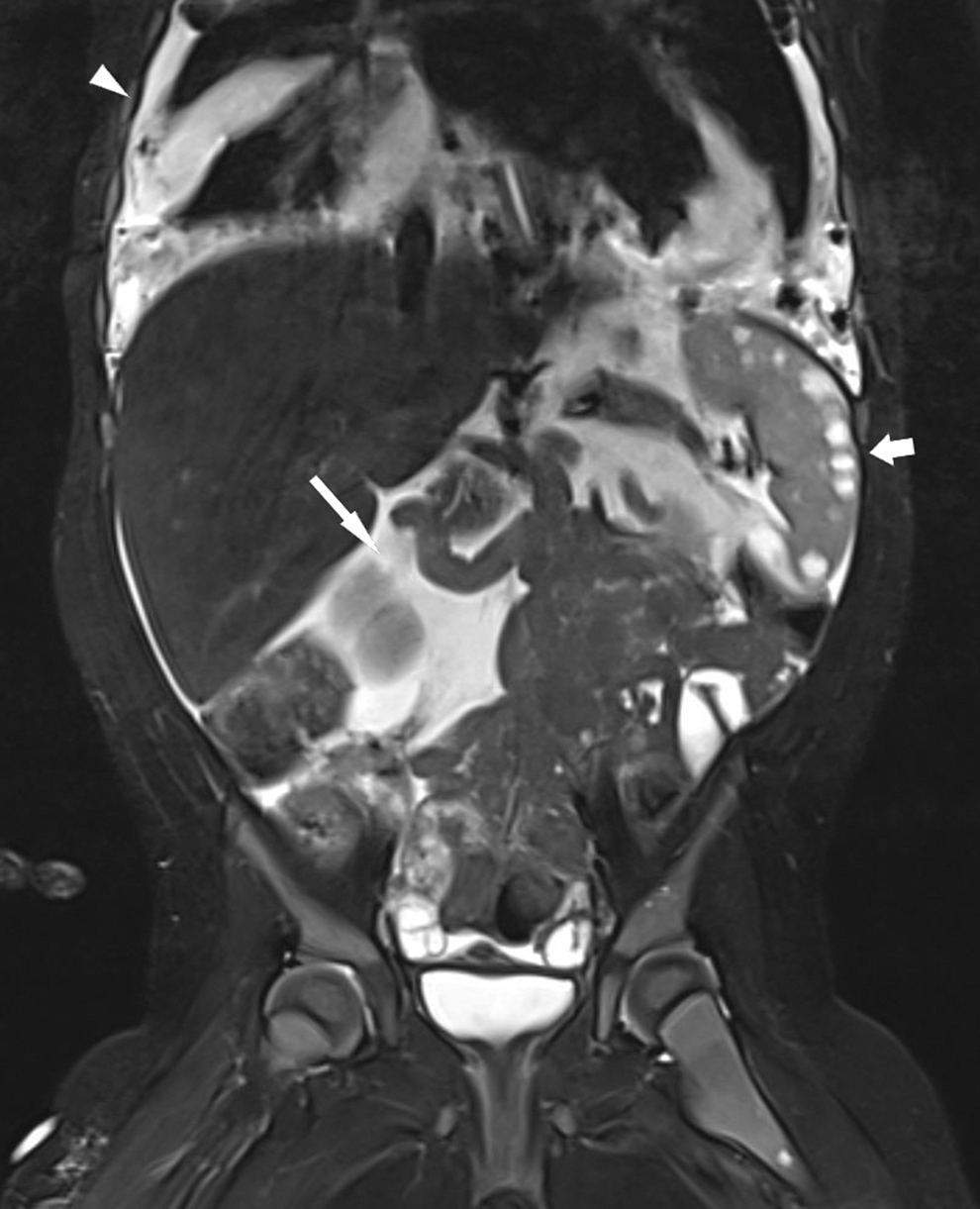
Radiological findings of a 4-year-old patient with GLA (see Table 2, patient 12). MRI coronary inversion recovery sequence: chylothorax (arrowhead), chylous ascites (arrow), and splenic lesions (short arrow). The patient exhibited multiple soft-tissue LMs (in the neck, mediastinum, and abdomen), organ involvement (lungs, liver, spleen, and ovaries), chylous effusions (ascites, pleural, and pericardial effusion), and bone lesions. Despite arrest of disease progression under multimodal treatment (e.g., pleural and peritoneal drainage, sirolimus, steroids, octreotide, and MCT diet), she was repeatedly hospitalized due to recurrent episodes of respiratory distress and respiratory tract infections, and eventually, a fatal episode of severe pneumonia with empyema and respiratory failure. MCT, medium-chain triglycerides.
Discussion
To our knowledge, we present the third largest series of CLA patients,5,36 and for the first time in the literature, a case series that is inclusive of all four clinical entities currently considered under the term “CLA.”
The name “GLA/GSD Registry” was coined before the term “CLA” was established, and the discriminatory features of the different clinical entities were described. Nonetheless, patients with all kinds of CLA participated in the study, and the latest diagnostic guidelines proposed by expert opinion consensus were retrospectively applied to the entire case series. 35
Overlapping features of CLA often make diagnosis challenging. Evidence of cortical bone destruction is a key aspect of GSD; spindled endothelial cells in histology are pathognomonic of KLA. One of our patients had both findings, and a GSD diagnosis was reached by consensus. Four patients were clinically diagnosed with CCLA, and the diagnosis was confirmed using MRL. 3 Another four patients with a diagnosis other than CCLA (2 GLA, 1 GSD, and 1 KLA) also had findings compatible with CCLA on MRL. This supports the suggestion that CCLA may actually be a part of GLA, GSD, or KLA. 37
In agreement with previous reports, pericardial effusion (n = 3) was a complication observed only in patients with pleural effusion, 12 and periosseous soft-tissue infiltration was associated with GSD.5,9,36 In previously published case series, relative sparing of the appendicular skeleton has been found in either GSD or GLA.5,36 In our case series, no such discrepancy was found.
The incidence of visceral involvement and the number of affected bones have been reported to be higher in GLA, 36 whereas progressive bone lesions and fractures are considered to be more common in GSD.9,36 We could not confirm these associations at a statistically significant level; however, a limitation of our study is that GLA and KLA patients were grouped for comparisons regarding osseous involvement, because of the small number of KLA patients.
As far as the diagnostic procedure is concerned, many patients did not undergo biopsy for various reasons (e.g., otherwise established diagnosis, contraindication, refusal, etc.). Since the identification of spindle cells in histology is a diagnostic criterion for KLA, 16 the lack of histological characterization renders misclassification between GLA and KLA possible. Given the inherent risks of tissue biopsy in CLA patients (bleeding, lymphorrhea, etc.), emerging noninvasive tests, such as liquid biopsy (i.e., New Generation Sequencing from cell-free DNA extracted from blood samples taken as close to the vascular malformation as possible) or single-cell RNA transcriptome sequencing, may prove to be helpful diagnostic tools.38–40
Regarding the genetic background of CLA, in many patients a somatic mutation is considered to be the likely cause. PIK3CA mutations were first discovered in GLA. 41 Recently, a somatic activating KRAS mutation was identified in tissue samples of patients with GSD, suggesting that targeted treatment with the MEK inhibitor trametinib could be effective.42,43 Furthermore, mutations in the NRAS proto-oncogene have been found in GLA and KLA.24,44,45 A mutation in CBL was recently identified in a patient with KLA. 46
Finally, in CCLA, apart from a somatic activating mutation in ARAF, a germline mutation in EPHB4 has been identified.33,34 However, none of these were known when our registry was set up; therefore, genetic testing was not part of the standard clinical care at the time. While we did identify two patients with a mutation likely causative of the CLA phenotype, larger numbers of patients are needed to characterize the genetics of CLA.
In the absence of standardized prospective follow-up data collection, this retrospective study was not designed to assess disease course or treatment outcomes. Interestingly, although KLA is known to have the most aggressive clinical course,15,16 an infant who was clinically diagnosed with KLA at birth exhibited spontaneous regression within a few months (Fig. 4). Thoracic involvement in CLA is associated with a poorer prognosis; in the case series presented by Ozeki et al 36 all documented deaths occurred in patients with thoracic involvement. Similarly, the only documented fatal outcome in our case series was indeed a 4-year-old GLA patient with thoracic involvement (Fig. 5).
Conclusions
In this retrospective observational study, patient data from a monocentric CLA registry were analyzed and presented as a case series. When it comes to such rare diseases, patient registries with standardized data collection are necessary to facilitate further research. This study demonstrates the frequency and features of principal symptoms and complications of CLA, as well as the extent of clinical overlap of these diseases, thereby highlighting the diagnostic challenges that clinicians face, even in specialized centers.
Although disease onset for the vast majority of our patients occurred in the first two decades of life, as has been previously described,5,36 in some cases the diagnosis was established up to decades later, underlining the importance of a high index of suspicion; CLA should be considered in the differential diagnosis in patients with LM combined with osteolysis, chylous effusions, or PLE. 35
Increased disease burden combined with limited response to treatment underpins the need for further understanding of CLA pathogenesis. It is probable that future discoveries in the field of underlying genetics of vascular malformations will lead to a new classification system based on common pathogenetic mechanisms. Regarding targeted treatment, several reports have described the treatment algorithms of different institutions; evidence-based treatment guidelines (e.g., indications for initiation, dosing, monitoring, and termination of treatment) still need to be established to optimize medical care. 3
Footnotes
Acknowledgments
This work was supported in part by the Center for Vascular Anomalies, Freiburg Center for Rare Diseases.
S.B., A.H., M.A., M.O., E.F., C.N., J.R., and F.K. are members of the Vascular Anomalies Working Group (VASCA WG) of the European Reference Network for Rare Multisystemic Vascular Diseases (VASCERN)—Project ID: 769036.
We thank the patients, their families, as well as their referring physicians, for supporting the registry. We also thank Susanne Forster for her support as a registry coordinator. Finally, we thank the LGD Alliance Europe, for constantly supporting our research activities in CLA. We thank PD Dr. C. Pieper (Department of Diagnostic and Interventional Radiology, University of Bonn) for providing examples of MR-lymhpangiography imaging.
Authors' Contributions
T.A.A. contributed to investigation (equal); methodology (lead); formal analysis (lead); writing—original draft (lead); writing—review and editing (lead); visualization (lead). S.B. assisted with resources (equal); writing—original draft (supporting). A.H. contributed to writing—review and editing (supporting). M.A. performed investigation (equal). M.O., E.F., and C.N. provided resources (equal). I.B. conducted funding acquisition (lead); writing—review, supervision and editing (supporting). J.R. and F.K. contributed to conceptualization (equal); project administration (equal); resources (equal); writing—review, supervision and editing (lead).
Author Disclosure Statement
T.A.A., J.R., and F.K. are investigators in the EPIK-P2 study (![]() ), sponsored by Novartis. F.K. reports receiving consulting fees from Novartis outside of the submitted work. J.R. is currently an employee of Novartis (since Nov. 2022). The other authors have no actual or potential conflicts of interest to disclose in relation to this publication.
), sponsored by Novartis. F.K. reports receiving consulting fees from Novartis outside of the submitted work. J.R. is currently an employee of Novartis (since Nov. 2022). The other authors have no actual or potential conflicts of interest to disclose in relation to this publication.
Funding Information
I.B. (Principal Investigator), J.R. (Coinvestigator), and T.A.A. (PhD student) were supported by the Swiss National Research Foundation, Sinergia grant CRSII5_1936942021-2024.