
Abstract
Canine distemper (CD) is a highly contagious, systemic, viral disease of dogs seen worldwide. The nucleocapsid protein (NP) of canine distemper virus encloses virus assembly and has some regulatory functions in viral transcription and replication. Here, we describe a procedure to generate a monoclonal antibody (MAb) against CDV N protein and investigate its characteristics. Western blot analysis showed that the MAbs produced in this study were against CDV N specifically. Indirect immunofluorescence assay demonstrated that they could recognize native N protein in CDV-infected Vero cells. The MAbs reported here may provide valuable tools for the further exploration of biological properties and functions of N protein and may also be developed for potential clinical applications.
Introduction
C
CDV belongs to a member of Morbillivirus genus in the Paramyxoviridae family. It possesses a single-strand, negative sense, approximately 13 Kb RNA genome that contains a long, open-reading frame. It has a non-segmented negative single-stranded RNA genome that codes for six structural proteins—nucleocapsid protein (NP), phospho protein (P), matrix protein (M), fusion protein (F), hemagglutinin protein (H), and large protein (L).
Nucleocapsid protein (NP) is the most abundant viral protein and the main regulator of the virus replication and transcription.(2) The nucleocapsid protein of canine distemper virus has three regions, the variable N-terminus, the variable C-terminus, and the highly conserved middle region.(3,4)
Previous studies showed that the middle region of NP (160–350 amino acids) is highly conserved among all the morbilliviruses and is fundamental for the function of the protein.(2) The recombinant protein, containing the conserved middle region of NP, shows good reactivity with the CDV antigen.(5) However, the functional role of the conserved middle region of NP in replication, virus infectivity, and induction of neutralizing activity is not fully understood.
In order to develop a useful tool for studying the conserved middle region of NP in canine distemper virus, for the present study we have generated a hybridoma cell line secreting monoclonal antibodies (MAbs) against the CDV N protein conserved middle region and have investigated some of its characteristics.
Materials and Methods
Cell lines, virus, and other reagents
African Green monkey kidney (Vero) cells and SP2/0 myeloma cells were cultured in RPMI-1640 medium (Hyclone, Beijing, China) supplemented with 10% fetal calf serum (Hyclone) and antibiotics (100 IU/mL ampicillin and 80 IU/mL streptomycin). All cells were maintained in a humidified 5% CO2 atmosphere at 37°C. The CDV strain CDV3 was propagated in Vero cells to prepare the antigen for WB and immunofluorescence assays.
Animals
Eight-week-old female BALB/c mice were purchased from the Experimental Animal Center of Jilin University (Changchun, China). The experiments that used animals were approved by the Animal Ethics Committee of Jilin University.
Construction of recombinant plasmid containing N gene of CDV
Total RNA was extracted from CDV-infected Vero cells via TRIzol (Invitrogen, Carlsbad, CA), according to the standard protocol, and the cDNA was produced with Superscript II RNAse H(-) reverse transcriptase (Invitrogen). In order to amplify the middle region of the NP gene from the cDNA template, a pair of primers were designed according to the sequence of CDV3 strain (GenBank accession no. EF375619). The upstream primer was 5′-GAAACTATGTATCCGGCT-3′ and the downstream primer was 5′-TGACTCACTCCATTCGGA-3′. The PCR products were purified with the EasyPure PCR Purification Kit (Transgen Biotech, Beijing, China) and subsequently cloned into pEASY-E1 Expression Kit (Transgen Biotech). The resulting recombinant plasmid was confirmed by nucleotide sequencing.
Expression and purification of NP protein middle region
The confirmed recombinant plasmids were transformed into Escherichia coli BL21 (DE3) (Novagen, Darmstadt, Germany) cells for expression. Overnight cultures of the transformed cells were diluted 1:100 in 50 mL Luria-Bertani (LB) broth containing120 mg/mL ampicillin at 37°C. When OD600 reached 0.6, 1 mM IPTG was added to the broth to induce expression and the cells were incubated with agitation for a further 4 h. Thereafter, bacterial cells were removed from the growth medium by centrifugation at 5000 g for 10 min and lysed by sonication in cold phosphate-buffered saline (PBS; pH 7.4). To investigate whether the recombinant protein was expressed as soluble or inclusion body, sodium dodecylsulfate-polyarcylamide gel electrophoresis (SDS-PAGE, 12%) was performed. Results showed that the His-tagged protein expressed predominantly as an inclusion body. To prepare antigen for immunization, we purified fusion protein by a His·Bind® Kit (Novagen). Briefly, the E. coli pellet obtained from the “large scale expression” was resuspended in 200 mL of ice-cold binding buffer. The cell suspension was sonicated on ice for 30 min. Inclusion bodies were collected by centrifugation at 5000 g for 15 min at 4°C, and subsequently resuspended in 100 mL binding buffer without denaturant followed by centrifugation at 5000 g for 15 min at 4°C. The resulting pellets were resuspended in 25 mL binding buffer containing 8 M urea. The suspension was incubated on ice for 1 h to completely dissolve the protein. Insoluble materials were removed by centrifugation at 16,000 g for 30 min at 4°C. The resulting supernatant was filtered through a 0.45 μm membrane and loaded into a His-bind resin column. When the prepared extract drained to the top of the column bed, the column was washed with 10 column volumes of binding buffer and 6 column volumes of wash buffer. Bound proteins were finally eluted with 2 column volumes of elute buffer. The purified His-NP was followed by analysis of SDS-PAGE in 12% polyacrylamide gel. Final concentrations of purified proteins were determined with a NanoDrop® ND-1000 Spectrophotometer (ThermoScientific).
Generation, selection, and purification of CDV-N specific MAb
Eight-week-old, female BALB/c mice were immunized subcutaneously with recombinant protein emulsified with Freund's complete adjuvant (Sigma, St. Louis, MO). The mice were then given two booster injections of purified protein emulsified with Freund's incomplete adjuvant. General MAb preparation procedures were performed as described previously with minor modifications. Splenocytes from immunized animals were harvested aseptically and fused to the myeloma cell line SP2/0 with polyethylene glycol 50% (w/v) PEG 4000 (Sigma) at a splenocyte-myeloma cell ratio of 5:1. The fused cells were cultured and selected in HAT medium (RPMI medium with 10% fetal bovine serum, 80 IU/mL streptomycin, 100 IU/mL ampicillin, 100 mM hypoxanthine,16 mM thymidine, and 400 mM aminopterin); aminopterin was omitted from the medium on the twelfth day. Cell culture supernatants from the surviving clones were screened by indirect enzyme-linked immunosorbent assay (ELISA), and positive cell lines were subcloned thrice by limiting dilution method. To produce large quantities of the MAbs, selected hybridoma cells were injected intraperitoneally into pristine-primed mice, and ascitic fluids were collected 10 days after injections. Isotype of the MAbs was determined with a mouse MAb isotyping kit according to the manufacturer's instructions (Sigma).
Western blot analysis
The MAb was analyzed by Western blot analysis with infected cells and recombinant proteins expressed in E. coli to determine their specificity and reactivity. Samples were mixed with an equal volume of sample loading buffer, boiled for 5 min, separated on 5% stacking/12% separating SDS polyacrylamide gels in a Tris-glycine buffer (0.025 M Tris base, 0.25 M glycine, 0.1% SDS), and transferred onto a nitrocellulose membrane with a Transblot apparatus (Bio-Rad, Hercules, CA). The membrane was washed three times in PBST with shaking, blocked with 2% BSA at 4°C overnight, and probed with MAb at 37°C for 1 h. Membranes were washed three times with PBST (0.1% Tween-20 in PBS) and incubated with anti-mouse IgG(H+L) HRP-conjugated secondary antibodies (Invitrogen) for 1 h. Membranes were washed three times with PBST, then protein bands were detected with DAB kit (CWBIO, Beijing, China).
Indirect immunofluorescence assays
Vero cells were cultured as monolayers and infected with CDV3. When 60–70% CPE was observed, cells were harvested and washed in PBS. Smears were coated with infect and mock-infect cells, air dried, and fixed with acetone. The MAb was added to glass slides as primary antibody. The slides were incubated for 1 h at 37°C in a moist chamber.
After washing three times with gentle shaking in PBS, 100 mL FITC-labeled goat anti-mouse IgG (Sigma), diluted 1:200 in PBS, was added and incubated at 37°C for 30 min. The smears were washed three times as described above, air dried, and mounted in glycerol. Finally, the cells were observed under the fluorescence microscopy.
Results
Construction of expression plasmid
The fragment of N gene corresponding to the middle region was amplified using the PCR (Fig. 1) and was then sequentially subjected to analysis with agarose gel electrophoresis to subclone into the pEASY-E1 vector. The recombinant plasmid pEASY-E1-N was transformed into E. coli BL21 (DE3) competent cells that were confirmed by sequencing.

PCR amplification the conserved middle regional N gene of CDV. M, DNA marker; lane 1, amplification product of the conserved middle regional N gene; lane 2, negative control.
Expression of conserved middle region of NP protein
To detect the expression of CDV middle region of NP protein in E. coli, the bacterial lysates were analyzed by SDS-PAGE. Results indicated that their recombinant protein was successfully expressed after IPTG induction, which mainly expressed in the form of an inclusion body. Western blot analysis results showed that the recombinant middle region of NP protein could be recognized by sera against CDV (Fig. 2).
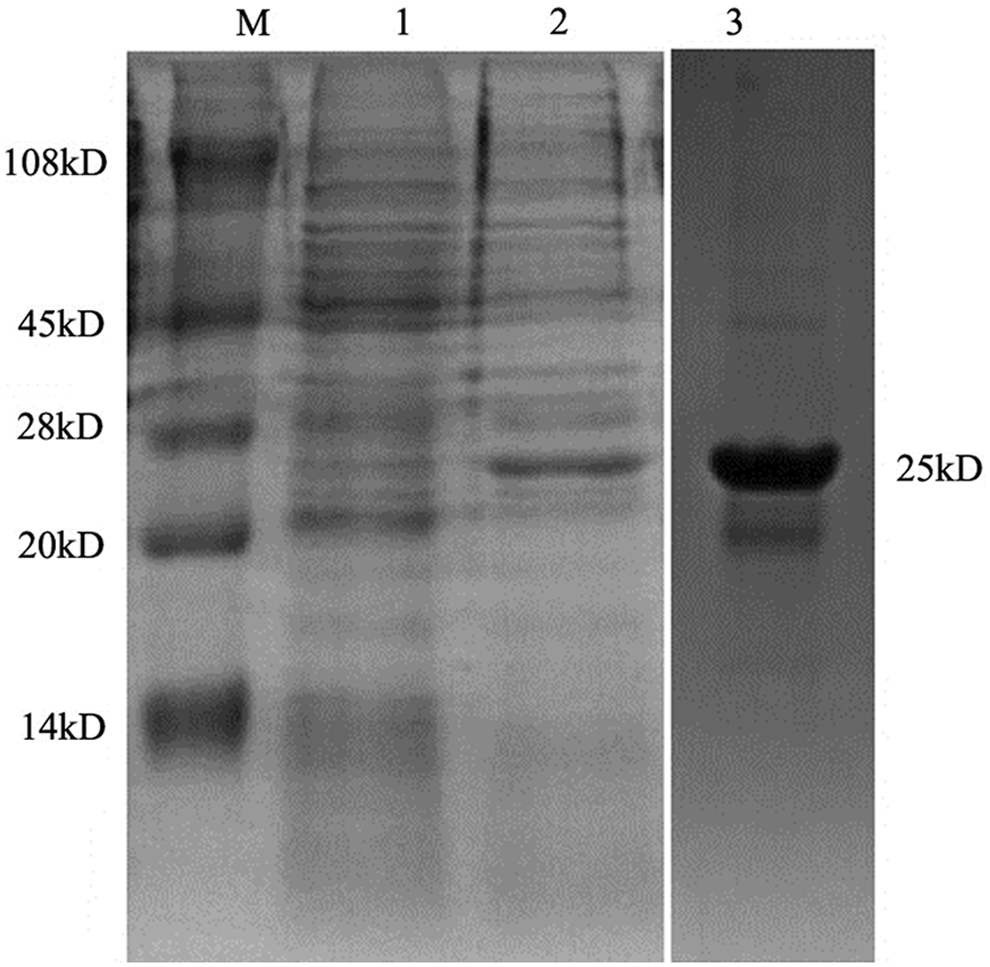
Expression analysis of CDV partial N protein. The bacteria harboring the recombinant plasmid were induced with IPTG and the bacterial protein was analyzed by SDS-PAGE. M, protein marker; lane 1, bacterial protein harboring CDV-N protein expressing plasmid but not induced with IPTG; lane 2, bacterial protein harboring CDV-N protein expressing plasmid; lane 3, purification recombination of CDV-N protein by His•Bind Kit.
Preparation of anti-CDV-N protein MAb
Five female BALB/c mice were immunized with the fusion protein. After three immunizations, the mouse that presented the highest antibody titer to the immunogen was selected and given a booster injection so that its spleen cells could be used in cell fusion. For screening with ELISA, three positive clones were obtained. After three rounds of subcloning, one strain showed strong and specific reactivity to CDV-N protein and was designated as 1N8. After isotyping, it was found that 1N8 was of IgG1 type, and the light chains were kappa type (Fig. 3).

Isotyping of monoclonal antibody. The isotype of MAb 1N8 was identified with mouse MAb isotyping kit.
Biological activity of MAb 1N8
Indirect ELISA was performed to measure the MAb titers. Results showed that the antibody titers of ascites were over 204,800 (Fig. 4). The specificity and reactivity were further determined by Western blot analysis using CDV-infected Vero cells. The results of Western blotting suggested that MAb 1N8 reacted with virus-infected cells (Fig. 5). These data revealed that the MAb has good specificity and reactivity. To identify the ability of specific MAbs to recognize the native N protein in CDV-infected cells, indirect immunofluorescence assay was carried out. A bright green signal could be detected, showing that the MAb recognized the native NP protein of CDV (Fig. 6).
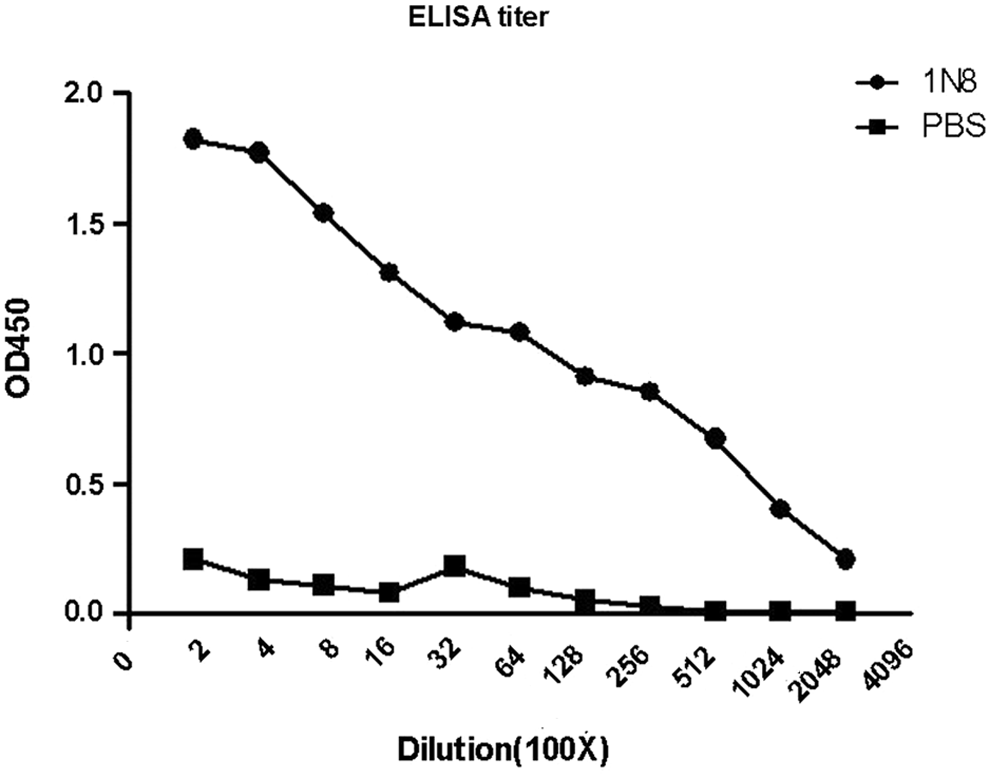
Titration of MAb 1N8 by ELISA. The ascites of MAb 1N8 was titered with indirect ELISA. PBS was used as the negative control. The dilution titer of MAb 1N8 ascites was over 204,800.
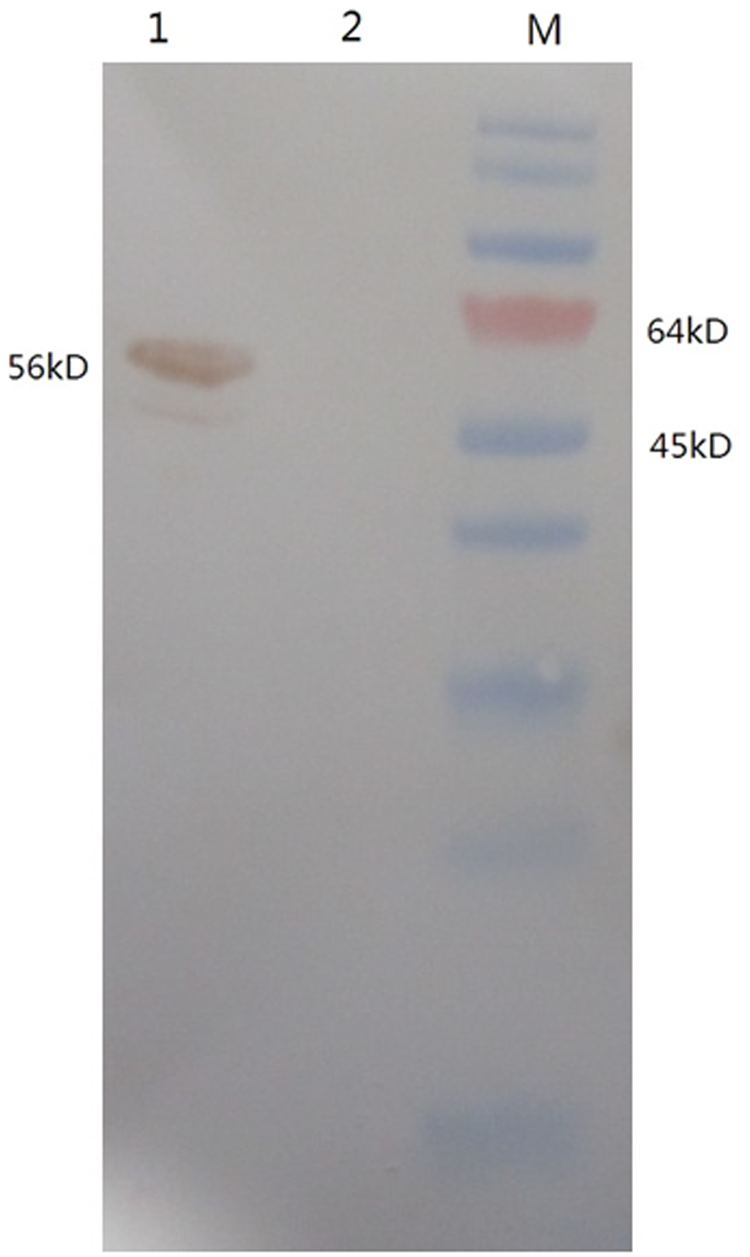
Western blot analysis shows the reaction to MAb 1N8 with CDV-infected Vero cells. Vero cells infected with CDV or mock infected were analyzed by SDS-PAGE and transferred to NC membrane, probed with the MAb 1N8, and developed with goat anti-mouse IgG. M, protein marker; lane 1, CDV-infected Vero cell lysate; lane 2, mock-infected cell lysate.
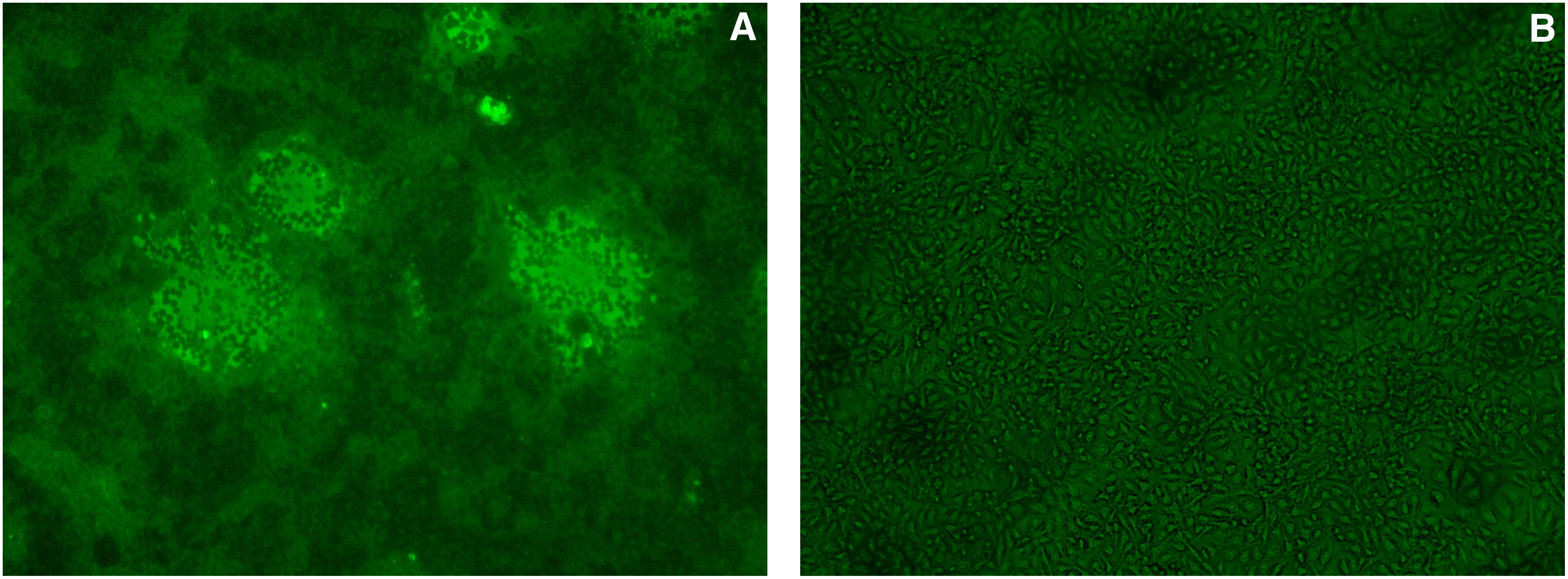
Indirect immunofluorescence assay analysis shows the reaction of MAb 1N8 with native NP protein on CDV-infected cells. (
Discussion
In this work, a monoclonal antibody against the CDVN protein was produced in BALB/c mice vaccinated with a middle region of NP protein expressed in E. coli. In previous studies, we have tried to express the full-length N protein in E. coli but at a considerably low level, possibly owing to over-length target fragment. Alternatively, the N protein was split into a middle region form to be expressed in E. coli, which has proven to be a better strategy than the previous one for production of MAbs.(6)
The middle region of NP protein, which is the conserved and functional region protein component of the CDV, was successfully expressed in E. coli using a prokaryotic expression vector pEASY-E1 containing a T7 lac promoter. In addition, a 6×His tag was introduced to the C-terminal ends of the middle region of NP protein. This strategy enhanced the integrity and affinity of our recombinant protein for Ni2+-agarose columns and increased the efficiency of protein purification.
One of the most powerful tools in the revelation of protein properties represents the use of MAbs, which have been successfully applied for clinical diagnosis and treatments.(7) There are few reports about the production of CDV N specific MAbs.
Previous reports describe that four monoclonal antibodies (MAbs) were established against the nucleocapsid protein (NP) of CDV by immunization with native virus. These investigators obtained MAbs that recognize the tertiary structure of the N-terminal region and linear epitopes in the C-terminal region of the CDV-NP.(7)
In this study, using a prokaryotic expressed recombinant protein as antigen to immunize the mice, we developed an MAb against CDV in the middle region of NP protein using the hybridoma technique. Our results indicated that the resulting MAb 1N8 recognized the N protein specifically.
Indirect immunofluorescence assay showed that the MAb could react with native N protein in CDV-infected Vero cells. We believe that the MAb generated in this study could be important for delineating undiscovered characteristics and functions of N protein, and may also be useful for clinical application in the diagnosis and therapeutics for controlling canine distemper.(8)
Footnotes
Acknowledgments
The present study was partly supported by the Ji Lin Province Major Scientific and Technological Achievements Transformation Program (no. 10ZDZH010) and the Ministry of Agriculture Scientific and Technological Achievements Transformation Program (no. 2010GB2B100098).
Author Disclosure Statement
The authors have no financial interests to disclose.