
Abstract
Recently Acinetobacter baumannii has emerged as a dominant form of nosocomial infections worldwide. As such, diagnostic tools are urgently needed. Reported here is the development/characterization of two mouse monoclonal antibodies (MAbs), F241G3sc2 and F241G6sc2, against A. baumannii ATCC 19606 with clinical strain cross-reactivity. Specifically, both MAbs cross-reacted with 33% of tested A. baumannii clinical strains, without cross-reactivity against other tested species of Acinetobacter. However, further testing with additional clinical strains and species is needed. Taken together, these results demonstrate both antibodies specifically target A. baumannii. With lower limits of detection at 0.32 ng/μL, both MAbs proved highly sensitive. Co-immunoprecipitation assays/LC-MS/MS, dot blots, and ELISAs eliminated the most abundant surface protein, outer membrane protein A (OmpA), as a protein target. However, since most of the proteins within the A. baumannii proteome are uncharacterized, exact protein targets could not be confirmed. Overall, these MAbs demonstrate practical applications, including ELISA, Western blot analysis, and co-IP assays, suitable to address the urgency for rapid detection tools required for A. baumannii research.
Introduction
A
Here we report the development and initial characterization of two MAbs, F241G3sc2 and F241G6sc2, specific and sensitive for the detection of A. baumannii. To our knowledge, these are the first MAbs to detect outer membrane proteins, which do not target the most abundant surface protein, outer membrane protein A (OmpA). Our MAbs are shown to serve practical applications, including ELISAs, Western blots, and co-immunoprecipitation assays, which may address the urgent need for A. baumannii detection tools.
Materials and Methods
Inoculation of mice and production of hybridomas and antibodies
Gamma-irradiated Acinetobacter baumannii 19606 (American Type Culture Collection, [ATCC], Rockville, MD) was utilized as the immunogen for development of MAbs within this study. Immunization of mice was performed according to the National Microbiology Laboratory (NML) standard operating procedures under ISO17025 in accordance with good animal practices outlined from the Canadian Council of Animal Care. Five- to six-week-old female BALB/c mice (Charles River, Wilmington, MA) were injected subcutaneously (s.c.) with 20 μg of gamma-irradiated A. baumannii 19606 in sterile phosphate-buffered saline (PBS, pH 7.4) emulsified with equal part Complete Freund's Adjuvant (H37-Ra, CFA, Difco, Oakeville, Canada) on day 0. The mice received four boosts of 20 μg of A. baumannii 19606 emulsified with Incomplete Freund's Adjuvant on days 28, 56, 84, and 112 and a final boost of 5 μg of A. baumannii 19606 in PBS 3 days prior to fusion. Mice were euthanized by anesthesia overdose, exsanguinated by cardiac puncture, and spleens removed using aseptic techniques. Hybridoma generation was conducted as previously described,(37) with the exception that Stemcell Clonacell Medium D (HAT, Vancouver, Canada) was supplemented with Roche hybridoma fusion cloning supplement (HFCS, Roche, Basel, Switzerland). MAbs subsequently underwent limiting dilution subcloning to ensure monoclonality. The MAb isotypes were determined using a mouse immunoglobulin isotyping kit according to manufacturer's recommended protocol (Roche). Supernatants from final clones were concentrated 10-fold using Amicon stirred cell nitrogen concentrators with Millipore YM-30 membrane (30 kDa, Millipore, Billerica, MA). MAbs were purified from the 10X serum-free media using a peristaltic pump and HiTrap protein G columns (GE Healthcare, Mississauga, Canada). Purified MAbs were dialyzed with 1× PBS pH 7.2 using Centriprep centrifugal filter units (Millipore) and concentrations determined using a BCA assay (Pierce, Rockford, IL).
Enzyme linked immunosorbent assay
End-point titer and hybridoma screening ELISAs were conducted as per the standard operating procedures of the Bioforensic Assay Development and Diagnostics, National Microbiology Laboratory, as previously described,(38) using 1000 ng/well of A. baumannii 19606 in PBS. Final values were background subtracted and 3x negative control.
Whole cell lysate preparations
A 500 mL culture of A. baumannii in TSY broth was grown for ∼16 h at 37°C. Bacterial cells were harvested by centrifugation at 3200 g for 30 min at 4°C using an Allegra X-12R centrifuge (Beckman Coulter, Mississauga, Canada). Supernatant was decanted and pellets stored at −80°C. Lysates were prepared per manufacturer's protocol using the Native Sample Prep kit (Invitrogen, Burlington, Canada). Protein quantity was determined using a BCA assay.
Outer membrane protein preparations
A. baumannii cells were streaked on a TSY plate and incubated ∼16 h at 37°C. A single colony was inoculated in 5 mL TSY broth and incubated ∼16 h at 37°C. The overnight culture was inoculated into 500 mL TSY broth and incubated ∼16 h at 37°C. Bacterial cells were harvested by centrifugation at 3200 g for 30 min at 4°C using an Allegra X-12R centrifuge (Beckman Coulter). Cells were resuspended in 20 mL 10 mM phosphate buffer (pH 6.0)/1 mM PMSF and sonicated at 50% power 5×30 s with 30 s cooling on ice in between. The cellular debris was removed by centrifugation at 6000 g for 10 min. The supernatant was removed by decanting and the pellet discarded. Supernatant was then centrifuged at 100,000 g for 2 h at 4°C using an Optima L-90K Ultra (Beckman Coulter). The pellets were resuspended in 4 mL 10 mM Tris followed by 16 mL N-Lauroyl sarcosine (protein-detergent ratio of 1:4). The mixture was incubated with gentle rocking for 30 min at room temperature (RT). OMPs were obtained by centrifugation at 50,000 g for 2 h at 15°C using an Optima L-90K Ultra centrifuge. Resulting pellets were washed with 50 mM Tris (pH 7.5) and resuspended in PBS (pH 7.2) and stored at −20°C.
Western blot analysis
Western blots were conducted per the standard operating procedures of the Bioforensic Assay Development and Diagnostics Section, National Microbiology Laboratory, as previously described.(38) The only changes involved loading 10 μL of A. baumannii 19606 whole cell extracts or 10 μL of OMP preparations isolated from A. baumannii 19606 per lane.
Immunodot blot cross-reactivity analysis
Bacterial strains Pseudomonas aeriginosa PAK14, Gamella haemolysins, Burkhoderia cepacia, Escherichia coli 25922 ATCC, A. baumannii clinical isolates ACBA-1 (urine), ACBA-2 (sacral wound), ACBA-3 (trachea), ACBA-4 (central line tip), ACBA-5 (Lt amputation), ACBA-6 (Lt inner thigh), and A. haemolyticus were spotted onto nitrocellulose membrane at a concentration of 1×107 cfu/mL. Recombinant OMP A (rOMPA) protein and BSA were also spotted at a concentration of 1.0 mg/mL. Membranes were blocked in 10% skim milk for 1 h at RT and washed three times for 10 min with TBST. Primary monoclonal antibodies (MAb, 1 mg/mL) were diluted 1:2000 in 10% skim milk and incubated with the membrane overnight at 4°C. Membranes were washed as described above. Goat anti-mouse-HRP secondary antibody was diluted 1:2000 (Southern Biotech, Birmingham, AL) in 10% skim milk and incubated for 1 h at RT. Positive binding was detected using DAB substrate.
Co-immunoprecipitation assay/protein identification by mass spectrometry
MAb F241G3sc2 or F241G6sc2 (100 μg) and corresponding isotype control antibodies F331G2sc2 or F09G3sc2 (100 μg), respectively, were mixed with 1 mg of A. baumannii whole cell lysate and incubated end-over-end at 4°C overnight. Subsequently, 100 μL of Protein A/G agarose resin (Pierce) was added to each mixture and incubated end-over-end at 4°C for 2 h. Antibody bound resin was precipitated by 1000 g centrifugation for 1 min followed by 3×1 mL Tris-buffered saline (pH 7.6)/0.1% Tween-20 (TBST) washes. Pellets were resuspended in 150 μL 4x Laemmli sample buffer and boiled for 5 min. Immunoprecipitated proteins (50 μL) were resolved by SDS-PAGE on 4–20% gradient Criterion gels at 150 V for 100 min. Proteins were visualized by staining using a Colloidal Blue Stain kit (Invitrogen). Individual protein bands and the corresponding region in isotype control lanes were excised using a scalpel blade and stored in ddH2O at −20°C. All protein extraction and mass spectrometry work was performed at the University of Victoria Genome BC Proteomics Center (Victoria, Canada) in accordance with in-house protocols. Trypsin digests were performed with a Genomic Solutions Pro-gest (DigiLab, Holliston, MA), as previously described by Parker and colleagues.(39,40) Briefly, 1 mm cube gel slices were cut, transferred to a Genomics Solutions Pro-gest perforated digestion tray, de-stained (50/45/5 v/v methanol/water/acetic acid), reduced (10 mM dithiothreitol; Sigma, St. Louis, MO), and alkylated (100 mM iodoacetamide; Sigma). Modified sequencing grade porcine trypsin solution (20 ng/μL, Promega, Madison, WI) was added to the gel slices at a ratio of 1:50 enzyme-protein and proteins digested for 5 h at 37°C. Tryptic digests and acid extraction of the gel slices (50/40/10 v/v acetonitrile/water/formic acid) were collected. Samples were then lyophilized and stored at −80°C prior to LC/MS analysis.
Peptide mixtures were separated by on-line reverse phase chromatography using a EASY-nLC II system (Thermo Fisher Scientific, Bremen, Germany) with a reversed-phase Magic C-18AQ pre-column (100 μm I.D., 2 cm length, 5 μm, 100 Å; Michrom BioResources, Auburn, CA) and reverse phase nano-analytical column Magic C-18AQ (75 μm I.D., 15 cm length, 5 μm, 100Å; Michrom BioResources) both prepared in-house, at a flow rate of 300 nL/min. The chromatography system was coupled on-line with an LTQ Orbitrap Velos mass spectrometer equipped with a Nanospray Flex source (Thermo Fisher Scientific). Solvents were A: 2% acetonitrile, 0.1% formic acid; B: 90% acetonitrile, 0.1% formic acid. After a 249 bar (∼10 μL) pre-column equilibration and 249 bar (∼6 μL) nanocolumn equilibration, samples were separated by a 55 min gradient (0 min, 5%B; 45 min, 45%B; 2 min, 80%B; hold 8 min, 80%B).
The LTQ Orbitrap Velos (Thermo Fisher Scientific) parameters were as follows: nano-electrospray ion source with spray voltage 2.1 kV, capillary temperature 225°C. Survey MS1 scan m/z range 400–2000 profile mode, resolution 60,000 FWHM@400m/z with AGC target 1E6, and one microscan with maximum inject time 500 ms. Lock mass Siloxane 445.120024 for internal calibration with preview mode for FTMS master scans: on, injection waveforms: on, monoisotopic precursor selection: on; rejection of charge state: 1. The ten most intense ions charge state 2–4 exceeding 5000 counts were selected for CID ion trap MSMS fragmentation (ITMS scans 2–11) with detection in centroid mode. Dynamic exclusion settings were: repeat count: 2; repeat duration: 15 s; exclusion list size: 500; exclusion duration: 60 s with a 10 ppm mass window. The CID activation isolation window was: 2 Da; AGC target: 1E4; maximum inject time: 100 ms; activation time: 10 ms; activation Q: 0.250; and normalized collision energy 35%. Common human keratin and porcine trypsin peptide masses were excluded from MS/MS selection during the analysis.
Raw files were analyzed with Proteome Discoverer 1.3.0.339 software suite (Thermo Scientific). Parameters for the Spectrum Selection to generate peak lists of the CID spectra (activation type: CID; s/n cut-off: 1.5; total intensity threshold: 0; minimum peak count: 1; precursor mass: 350–5000 Da). The peak lists were submitted to an in-house Mascot 2.2 server against a Uniprot-Swissprot 20121115 (538,259 sequences; 191,113,170 residues) with taxonomy other Proteobacteria (153,160 sequences).
Database search parameters were as follows: precursor tolerance 8 ppm; MS/MS tolerance 0.6 Da; Trypsin enzyme 1 missed cleavages; FT-ICR instrument type; fixed modification: carbamidomethylation (C); variable modifications: deamidation (N,Q), oxidation (M), Propionamide (C). The Decoy database Percolator settings: Max delta Cn 0.05; Target FDR strict 0.01, Target FDR relaxed 0.05 with validation based on q-value.
Results
Mice were immunized with A. baumannii strain ATCC 19606 whole bacteria. Spleens from immunized mice, sero-positive against A. baumannii whole bacteria, were used in two separate hybridoma fusions that generated 1288 clones. Positive clones (91) were selected on the basis of producing MAbs specifically against A. baumannii in a primary ELISA screen. Subsequent secondary ELISA screening isolated a total of 2 clones as true positives for producing anti-A. baumannii MAbs. End-point ELISA titers for F241G3sc2 and F241G6sc2 were found to be 125 ng/mL and 0.8 ng/mL, respectively, against whole bacteria. The fusion, isotype, and initial characterization of the panel of generated MAbs are shown in Table 1. The two MAbs were of isotype IgG1/κ (F241G3sc2) and IgG2a/κ (F241G6sc2).
A. baum, Acinetobacter baumannii; A. haem, Acinetobacter haemolyticus; A. cal, Acinetobacter calcoaceticus; A. john, Acinetobacter johnsonii.
ELISA results using whole A. baumannii bacteria as capture antigen determined a lower limit of detection at 0.32 ng/μL for both F241G3sc2 and F241G6sc2.
Both MAbs were examined for cross-reactivity to different Acinetobacter species and clinical isolates of A. baumannii using immunodot blot and ELISA assays. The results are presented in Figure 1A and summarized in Table 1. Both MAbs were positive (+) against A. baumannii 19606 in both immunodot blots and ELISAs. Cross-reactivity was negative (−) for other Acinetobacter species (Table 1) and positive for 2/6 clinical isolates assayed (Fig. 1A). In addition, neither F241G3sc2 nor F241G6sc2 binding to recombinant OmpA (rOmpA) or BSA (negative control) was detected.
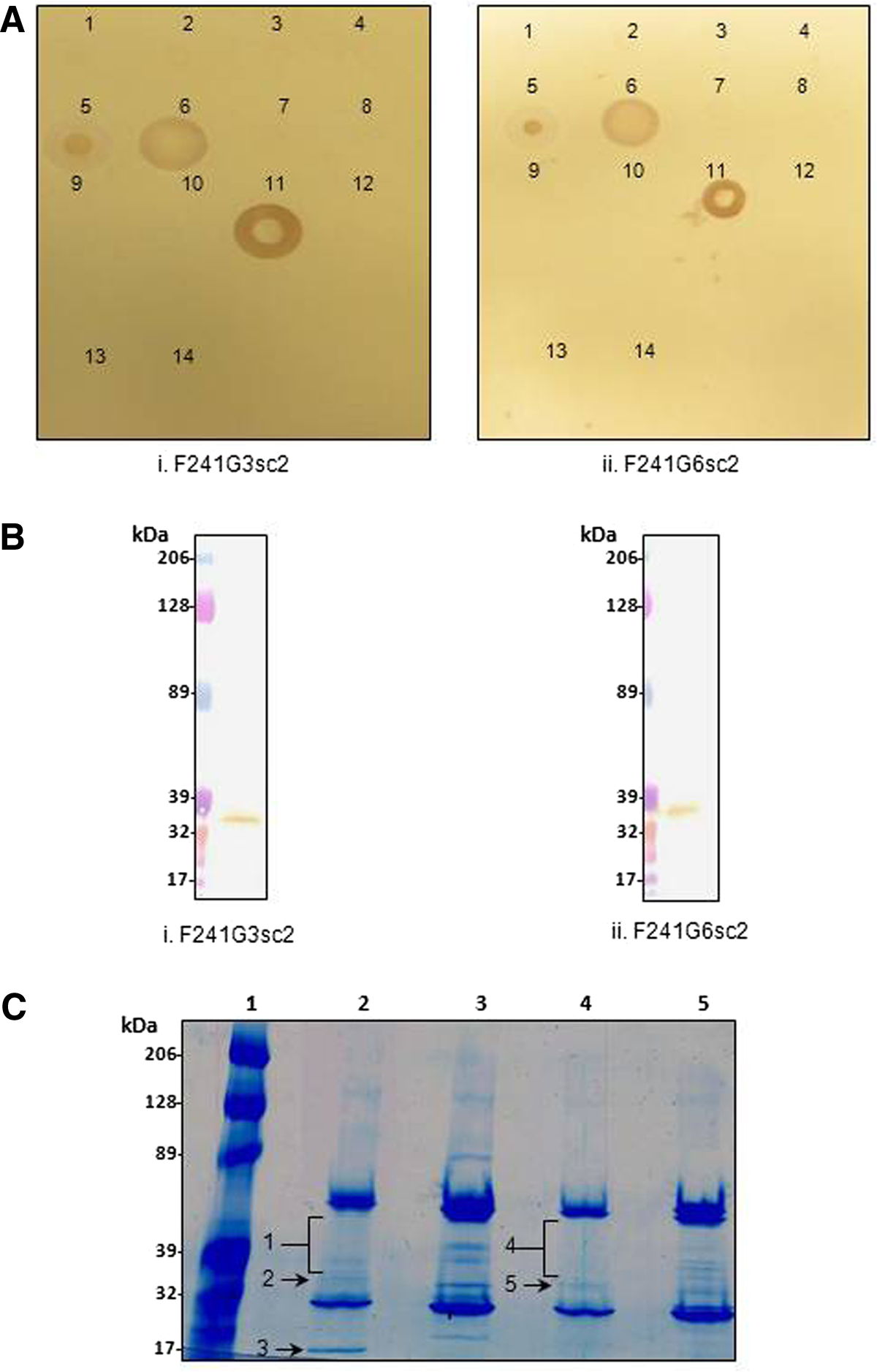
Species and protein targets of anti-A. baumannii MAbs. (
To further elucidate practical applications for F241G3sc2 and F241G6sc2, Western blot analyses were conducted. As seen in Figure 1B, both antibodies were positive under the specified conditions (see Western blot methods). Furthermore, both F241G3sc2 and F241G6sc2 detected a single protein band. Although the exact molecular masses of each protein band were not determined, they are estimated at ∼35 kDa. It is possible that the protein band for F241G3sc2 has a lower molecular weight than that of F241G6sc2. Negative control isotype antibodies were simultaneously assayed and did not detect any protein bands in Western blots (data not shown).
Co-immunoprecipitation (co-IP) assays were performed to identify the antigenic target proteins for both F241G3sc2 and F241G6sc2 detected in Western blots (Fig. 1B). As seen in Figure 1C, various protein bands ranging from ∼25–50 kDa and ∼18–50 kDa were resolved for F241G3sc2 and F241G6sc2, respectively. Due to the various bands being compressed within a tight range of ∼25 kDa (excluding the ∼18 kDa protein band in lane 2), proteins were divided in two. A single protein band (2, 3, and 5) was excised or a large region (bands 1 and 4) from the colloidal blue stained gel (Fig. 1C). The corresponding regions on the negative control isotype lanes were also excised. Proteins common between the negative control isotype and the experimental MAb lanes were dismissed along with all intracellular proteins (data not shown).
To be selected, MAb antigenic protein(s) had to meet the following criteria: expressed in A. baumannii (ideally membrane subcellular localization); MOWSE score of >40; theoretical molecular weight corresponding to that of the protein band seen in the colloidal blue-stained gel; at least two peptides; and a minimum peptide length of eight amino acids. Antigenic target protein identifications based on these criteria are presented in Table 2. Despite strict screening conditions, multiple target proteins qualified for F241G6sc2, whereas only a single potential target was identified for F241G3sc2 (34 kDa outer membrane protein). As seen in Table 2, there are four proteins identified as putative uncharacterized protein. Unique SwissProt Identifiers do not necessarily translate to individual proteins; however, based on primary sequence analysis, these are all in fact different proteins. Furthermore, peptides used to identify each protein were mutually exclusive. Finally, no proteins in band 4 were identified that met the criteria for selection.
Discussion
Although A. baumannii poses little risk to healthy individuals, patients with open wounds, on mechanical ventilation, or those with chronic diseases are highly susceptible to infections.(18) As a major cause of nosocomial infections, A. baumannii is attributed with significant morbidity and mortality rates within hospitals, especially among patients in intensive care units (ICU)(13) and immunosuppressed individuals.(11,15) Since A. baumannii can easily spread to susceptible individuals through common modes of transmission, such as person-to-person contact or environmental exposure, rapid detection and identification of the bacterium are of tremendous value. If A. baumannii can be detected prior to susceptible individuals becoming exposed, perhaps infections can be reduced. This preventative approach of pre-exposure detection might better serve in inhibiting infections rather than attempting to treat highly resistant strains post-exposure for which no therapy is currently available. Pre-exposure detection is only possible with the aid of highly sensitive and specific reagents, such as antibodies.
MAbs offer multiple unique advantages, such as increased specificity, unlimited availability, and consistent properties, which are ideal for research and detection applications.(40,41) Here we report the development and characterization of two mouse MAbs, F241G3sc2 and F241G6sc2, for the detection of A. baumannii. Whole A. baumannii bacteria were used for immunization to generate hybridoma clones. This immunization approach allows for the generation of antibodies that recognize antigens with the same conformation as the target protein expressed on surface of the bacteria leading to greater sensitivity. This is illustrated by ELISA results, which demonstrated a lower limit of detection at 0.32 ng/μL for both F241G3sc2 and F241G6sc2. As such, both MAbs proved highly sensitive against the bacteria (Table 1).
Both antibodies bound 33% (2/6) clinical isolates of A. baumannii. This finding is not surprising. Bacterial infections within patients undergo selective pressure, resulting in genetic and/or phenotypic changes.(42) This is readily apparent in the emergence and evolution of antibiotic resistance.(43) Furthermore, McCarthy and colleagues have shown that Staphylococcus aureus undergoes genetic variation within surface expressed proteins as a means to evade host immunity.(44) The authors also concluded that changes in the surface proteins do not affect virulence.(44) Accordingly, clinical isolates of A. baumannii may also undergo changes in surface proteins, which alter antigenic sites, thereby inhibiting binding of F241G3sc2 and F241G6sc2 antibodies. As such, the percentage of clinical strain cross-reactivity for MAbs targeting a cultured bacterial strain cannot be regulated or predetermined. Only six clinical strains were available for this study and in turn assayed for cross-reactivity. It is therefore possible that, when additional clinical strains become available, more will be identified that are bound by F241G3sc2 and/or F241G6sc2.
In addition, regarding specificity, neither F241G3sc2 nor F241G6sc2 was found to cross-react with any of the other three strains of Acinetobacter (A. johnsonii, A. haemolyticus, or A. calcoaceticus) tested using either dot blots (Fig. 1A) or ELISA assays (Table 1). These two independent findings demonstrate that both MAbs are specific for A. baumannii and not other species in the Acinetobacter genus. This is relevant as A. baumannii accounts for 80% of Acinetobacter infections(6) and therefore, specificity is essential.
Of further importance is the finding that both F241G3sc2 and F241G6sc2 detected A. baumannii whole cell and/or whole cell lysate in ELISA and Western blot assays. Both assays are essential in bacterial detection and identification. These findings show F241G3sc2 and F241G6sc2 are suitable for routine assays, thereby demonstrating practical applications of these two antibodies.
Interestingly, both MAbs detected target proteins under reduced and denatured (i.e., SDS-PAGE) conditions (Fig. 1B). This in turn suggests the antibodies may be independent of conformation and only target linear epitopes. Furthermore, due to the slight differences in the molecular masses of target proteins (Fig. 1B), along with different identifications by LC-MS/MS (Table 2), the two antibodies most likely bind different target proteins. Taken together, the results suggest two distinct target molecules.
Although the genome of A. baumannii has been sequenced, most proteins remain uncharacterized. As such, extensive proteomic studies are required before A. baumannii proteins can be identified and MAb targets confirmed by independent assays. Co-IP LC-MS/MS results identified various proteins as potential targets for F241G3sc2 and F241G6sc2 (Fig. 1C). Albeit some of the proteins listed in Table 2 correspond to the molecular weight of the protein bands detected in Western blots (Fig. 1B), some do not. For example, band 1 (Fig. 1C) has a molecular mass larger than the expected ∼35 kDa band (Fig. 1B), whereas band 3 (Fig. 1C) has a lower molecular mass. From this preliminary study, 34 kDa outer membrane protein and SPFH/band 7-like proteins may be the corresponding targets for F241G3sc2 and F241G6sc2, respectively. Both potential target proteins are of similar molecular mass to protein bands detected by Western blotting (Fig. 1B), the theoretical molecular weight matches the visible bands in colloidal blue-stained SDS-PAGE gels (Fig 1C), both proteins are expressed in A. baumannii and are located in the outer membrane of the bacterium. Furthermore, these proteins were not identified in the negative control isotype samples, suggesting specific interactions with the corresponding antibody. However, independent confirmation is still required once additional tools (recombinant proteins, additional antibodies) are developed. Despite corroborative evidence, these same experiments also yielded various putative uncharacterized proteins as potential targets. These uncharacterized proteins cannot be excluded because some are localized to the outer membrane while others are integral proteins. As seen from the MOWSE scores and minimum peptide lengths (Table 2), all proteins identified using MASCOT were significant and the results reproducible in two independent experiments. As such, the results cannot be disregarded as irrelevant or irreproducible. Overall, definitive target identification cannot proceed until further progress is made on the A. baumannii proteome.
Further studies are needed to assess the functional and therapeutic potential of both F241G3sc2 and F241G6sc2 against A. baumannii along with additional practical assays such as immunofluoresence and cytometry. Along with these studies and the development of additional reagents, confirmatory experiments will be conducted to determine the target protein and map the epitopes targeted by F241G3sc2 and F241G6sc2 on A. baumannii. Although the exact antigenic target proteins were not identified definitively, OmpA, frequently targeted by antibodies, was eliminated by co-IP/MS (Table 2), dot blot (Fig. 1A), and ELISA (data not shown). This in turn, demonstrates that both F241G3sc2 and F241G6sc2 bind outer membrane targets for which, to our knowledge, no MAbs are currently available.
Due to the ever-increasing cases of nosocomial A. baumannii infections, this short communication was compiled to inform researchers on the availability of F241G3sc2 and F241G6sc2 antibodies. This preliminary study has demonstrated the practical applications of these two novel MAbs, which may serve to help further studies on the identification and rapid detection of A. baumannii along with certain clinical strains. However, additional work is necessary to test both antibodies against a larger panel of both clinical strains and clinically relevant species. These data will strengthen the clinical relevance and the specificity of these antibodies.
Footnotes
Acknowledgments
The authors would like to thank Derek Smith and the University of Victoria Genome BC Proteomics Center for sample preparation and for performing all the LC-MS/MS work. In addition, we thank Dr. Andrew Simor at Sunnybrook Health Sciences Center (Toronto) for providing all A. baumannii clinical samples. This research is supported by the Public Health Agency, Government of Canada.
Author Disclosure Statement
The authors declare there are no conflicts of interest. The views and opinions expressed herein are those of the authors only and do not necessarily represent the views and opinions of the Public Health Agency of Canada or the Government of Canada.