
Abstract
Expression of transcripts for the homotypic adhesion protein epithelial V-like antigen 1 (EVA1), also known as myelin protein zero like-2 (Mpzl2), is known to be present in thymic stromal cells. However, protein expression within different thymic subsets, stromal and/or lymphoid, has not been characterized due a lack of specific reagents. To address this, we generated a hybridoma (G9P3-1) secreting a monoclonal antibody (G9P3-1Mab), reactive against both human and mouse EVA1. The G9P3-1Mab was generated by immunizing Mpzl2-deficient gene-targeted mice with the extracellular domain of EVA1, followed by a conventional hybridoma fusion protocol, illustrating the feasibility of using gene-targeted mice to generate monoclonal antibodies with multiple species cross-reactivity. We confirmed expression of EVA1 on cortical and medullary epithelial cell subsets and revealed a restricted pattern of expression on CD4- CD8- double negative (DN) cell subsets, with the highest level of expression on DN3 (CD44lowCD25+) thymocytes. G9P3-1MAb is a valuable reagent to study thymic T cell development and is likely useful for the analysis of pathological conditions affecting thymopoiesis, such as thymic involution caused by stress or aging.
Introduction
T
Thymic cortex and medulla are the two major non-hematopoietic stromal environments in the thymus. These are formed by characteristic subsets of epithelial cells that can be identified by their differential expression of cell surface markers, typically identified as cortical (Ly-51highUEA-1low) and medullar (Ly-51lowUEA-1high) thymic epithelial cells (TECs).(2) This extra layer of anatomical complexity implies a re-definition of thymocyte development in terms of the anatomical location of the developmental stages.(3) Along the same lines, early stages of thymocyte development occur at the inner cortex microenvironment close to the corticomedullary junction (CMJ), where early thymic progenitors begin to differentiate and migrate to the outer cortex to sequentially become DN2, DN3, and DP thymocytes. Following positive selection and CD4 or CD8 lineage decision, thymocytes migrate to the medulla to undergo negative selection and finalize their differentiation to become mature T cells ready to populate the peripheral lymphoid organs.(4–6)
In addition to providing an anatomical framework, TECs positively influence thymocyte development by delivering microenvironment-specific pro-developmental signals.(7,8) Conversely, developing thymocytes are active supporters of TEC differentiation, function, and survival.(9–12) Altogether this coined “lymphostromal” cross-talk is an essential feature for proper thymic function and, consequently, its disruption is associated with thymic dysfunction.(13)
Cell adhesion molecules play a key role in orchestrating the circulation of developing thymocytes through the cortical and medullar thymic areas and by supporting pro-developmental cross-talk interactions.(3) Several studies have described the importance of high affinity adhesion molecules in thymus function(14–17); however, studies addressing the role of low affinity adhesion molecules are scarce due in part to the experimental difficulties in identifying low avidity cell-cell interactions, rather than their lack of functional relevance.(18)
Epithelial V-like antigen 1 (EVA1; Mpzl2) is a low affinity homotypic adhesion type I transmembrane glycoprotein bearing an immunoglobulin V-type domain.(19,20) Previously published reports, and our own work, have demonstrated that EVA1 expression is strongly upregulated at the DN3 stage of thymocyte development.(21,22) In addition, EVA1 is highly expressed in cortical and medullar TECs and its transgenic overexpression in these cells cause important changes in thymic cellularity and structure.(23,24) Furthermore, a potential role for EVA1 in promoting cellular regeneration of involuted thymuses was proposed,(21) presumably by promoting pro-developmental DN3-cortical epithelial cell interactions.
In this report, we describe a new MAb against mouse and human EVA1, which allows for the identification of DN3 thymocytes as well as cortical and medullar thymic epithelial cells by flow cytometry. This antibody can potentially be used in studies of stress and age-dependent thymic involution and regeneration. Moreover, this new anti-EVA1 MAb may prove to be a valuable reagent for cell purification protocols aimed at deciphering the function of different thymocyte developmental subsets.
Materials and Methods
Generation of Eva1 knockout mouse
We conducted a standard immunization protocol using Mpzl2-deficient gene-targeted mice (unpublished data). Following standard gene targeting techniques in embryonic stem cells, we generated an Mpzl2 conditional knockout mouse in which exons 2 and 3 encoding for the extracellular portion of EVA1 were flanked by two loxp sequences (Eva1flox). This targeting strategy allowed us to study the role of EVA1 in a tissue-specific fashion with the use of the proper tissue specific Cre-deleter mouse strains, as well as to generate a full Eva1 knockout mouse for immunization and hybridoma generation.
To generate a full homozygous EVA1 knockout mouse (Eva1ko/ko) in which Mpzl2 is deleted on both chromosomes, we crossed homozygous Eva1flox/flox mice with the B6.C-Tg (CMV-cre) 1Cgn/J strain (Jackson Laboratory, Bar Harbor, ME), in which Cre is under the control of the human cytomegalovirus minimal promoter, affording Cre expression (and Mpzl2 gene deletion) in virtually all tissues, including germ cells. The F1 generation containing one Eva1 knockout allele (Eva1ko/wild type) was backcrossed 10 times to the C57BL/6 (B6) genetic background. Finally, we intercrossed Eva1ko/wild type mice to select those Eva1ko/ko for immunization.
Retroviral transduction of HEK-293T cells
MSCV-based retroviral constructs encoding the human and mouse EVA1 cDNA in tandem with an IRES-YFP reporter sequence (EVA1-YFP) were generated following conventional cloning techniques. We transfected HEK-293T cells with mouse and human EVA1-YFP along with gag-pol and VSV-G constructs, and 48 h later supernatants enriched with VSV-G pseudo-typed retroviral particles were harvested. Subsequently, concentrated supernatants were used to transduce HEK-293T cells by spin infection for 1 h at 2.7×103 rpm. Transduced HEK-293T-YFP+ cells, stably expressing human or mouse EVA1, were used to test our monoclonal antibodies by flow cytometry.
Purification of mouse extracellular-EVA1 protein fused to human IgG3 Fc portion
Following conventional cloning techniques, we generated a lentiviral construct encoding the extracellular portion of mouse EVA1 genetically fused to the Fc portion of human IgG3 (ext-Eva1-Fc) in tandem with an IRES-GFP reporter sequence. We then produced VSV-G pseudo-typed lentiviral particles that were used to transduce CHO-S cells (Invitrogen, Carlsbad, CA) by spin infection for 1 h at 2.7×103 rpm. Transduced CHO-S cells were FACS-sorted for high levels of GFP and expanded for protein production with Free-Style protein free medium (Invitrogen, Carlsbad, CA).
Growing GFP+ CHO-S cells were expanded in two 500 mL roller bottles (Corning, New York, NY) for a period of 7 to 8 days, cells were pelleted, and the ext-EVA1-Fc enriched supernatant harvested, concentrated, and dialized overnight. Finally, ext-EVA1-Fc protein was purified by FPLC (AKTA-GE Healthcare Life Sciences, Pittsburgh, PA) with a protein-G affinity column. The final purified solution was quantified for protein content. Protein purity was visually assessed in a 12% SDS-PAGE gel stained with Coomassie Brilliant Blue.
Immunization of mice
Eva1ko/ko mice were given an intraperitoneal injection of 100 μL (50 μg) of ext-EVA1-Fc protein, emulsioned with 100 μL of Sigma Adjuvant System 2x (Sigma-Aldrich, St. Louis, MO), on days 0 and 14, and with 25 μg on days 28 and 42. The final boost of 25 μg without adjuvant was given on day 56. On day 59, a selected mouse was killed and splenocytes used for fusion. We chose the best responder of three immunized mice based on the anti-ext-EVA1-Fc antibody titer of the serum extracted on day 21.
Fusion protocol and hybridoma screening by ELISA and flow cytometry
For the fusion 4×106 splenocytes and 40 million of the myeloma parental cell line P3X63Ag8.653 (ATCC, Manassas, VA) were used for a conventional PEG-1500 (Sigma-Aldrich) fusion protocol.(25) After fusion, cells were plated in eight 96-well plates and hybridomas selected with HAT (Sigma-Aldrich) for 2 weeks, followed by a transition culture with HT medium (Sigma-Aldrich) for an additional 2 weeks. We used RPMI media supplemented with glutamax (Gibco, Grand Island, NY), sodium pyruvate (Gibco), gentamycin (Gibco), BM Condimed H1 at 10% (Roche, Basel, Switzerland), and fetal bovine serum at 10% (Sigma-Aldrich) with or without HAT or HT. Supernatants from wells showing hybridoma growth were tested by ELISA. We coated 96-well ELISA plates with 5 μg/well of ext-EVA1-Fc protein in 100 μL of BBS buffer and left them to incubate at 4°C for 12 h. The next day, wells were blocked with PBS-BSA 10% for 1 h and washed. Subsequently, hybridoma supernatants (100 μL) were added to each well and incubated for 1 h at room temperature. Final steps included incubation with biotinylated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, PA) for 1 h RT, incubation with avidin-alkaline phosphatase for 1 h RT, incubation with p-nitrophenyl phosphate disodium hexahydrate substrate for 1 h, and reading in an ELISA plate reader at 405/650 nm. All washes between incubation steps were performed with PBS-Tween.
From the highest ELISA-positive hybridoma supernatants, we determined the reactivity against mouse and human EVA1 by flow cytometry. 1×106 HEK-293T-YFP+ cells expressing human or mouse Eva1 were incubated with 150 μL of hybridoma supernatant in a 5 mL FACS tube for 1 h at 4°C. Cells were washed twice with 5 mL of PBS-BSA 0.1% and incubated with biotinylated anti-mouse-IgG for 30 min, washed, and incubated with streptavidin-eFluor450. After a final wash, cells were resuspended in 500 μL of PBS and analyzed by flow cytometry.
Preparation and analysis of thymic epithelial cells by flow cytometry
We prepared a single cell suspension of mouse thymic epithelial cells following a previously published protocol.(26) Briefly, each individual thymus was cut into three or four pieces and enzymatically digested at 37°C with liberase (Roche) until a single cell suspension was obtained. We then depleted cells from the hematopoietic lineage (CD45+) using anti-CD45 coated magnetic MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer specifications. CD45-depleted cells were stained with G9P3-1 supernatant followed by anti-mouse-IgG-biotin (Jackson Immunoresearch) and streptavidin-eFluor450. Finally, cells were incubated with anti-CD45 APC (30-F11), anti-EpCAM PeCy7 (G8.8), anti-Ly-51 PE (BP-1), and UEA-1 FITC (Sigma-Aldrich). All washes between steps were performed twice with 4 mL of PBS-BSA 0.1%. Thymic epithelial cells were identified as CD45- EpCAM+ by flow cytometric analysis.
Preparation and analysis of thymocytes by flow cytometry
A thymus single cell suspension was prepared by mechanically smashing the thymus with two glass slides. Single cell suspension was collected, washed, and stained with G9P3-1 supernatant (500 μL) followed by anti-mouse-IgG biotin (Jackson Immunoresearch), streptavidin-eFluor450, anti-CD44 FITC (IM7), anti-CD25 PerCPCy5.5 (PC61.5), anti-CD28 APC (E18), anti-CD4 PE (GK1.5), and anti-CD8 PE (53-6.7). All washes between steps were performed twice with 4 mL of PBS-BSA 0.1%. DN thymocytes were identified as being CD4 and/or CD8 negative.
Cell sorting and real-time PCR
We FACS-sorted (FACSAria-BD Biosciences, San Jose, CA) 500 cells/well into a U-bottom 96-well plate from each one of following DN thymocyte populations: DN1 (CD44+CD25-), DN2 (CD44+CD25+), DN3 (CD44lowCD25+), DN4 (CD44-CD25-) as well as a pool of DP (CD4+CD8+) and SP (CD4+ and CD8+) thymocytes (DP/SP). Each well contained a dual reverse transcriptase/lysis solution with 5 mM DTT, 2 U/μL RNAase, 500 μM dNTPs, 10 U/μL of Superscript reverse transcriptase (Invitrogen), 100 μg/mL bovine serum albumin, 1% Triton X-100, 25 ng/μL Oligo dT (Invitrogen), 0.5 nM spermidine, and 1X First Strant buffer (Invitrogen) in a final volume of 20 μL. For cDNA synthesis, the plate was incubated 60 min at 50°C followed by a heat inactivation step of 15 min at 70°C. For real-time PCR, we used 1 μL of cDNA solution in a final volume of 12.5 μL Power SyBRGreen PCR master mix solution (Applied Biosystems, Foster City, CA). To quantify EVA1 transcripts, we used EVA1F (CTAACACTTCGAGAAGCTGAGGATT) and EVA1R (GGTCATCGCTGGAAACATTTC) primers. Normalization was performed against β-actin, which was amplified with β-actinF (GGCTCTTTTCCAGCCTTCCT) and β-actinR (GTCTTTACGGATGTCAACGTCACA) primers. Samples were run over 50 cycles with a 7900HT Fast Real Time PCR (Applied Biosystems).
Immunoglobulin isotyping
An IsoStrip isotyping kit (Roche) was used to determine the heavy and light chain isotype of our anti-EVA1 MAb.
Results
Purification of ext-EVA1-Fc protein
CHO-S cells were transduced to express ext-EVA1-Fc retroviral construct, and transduced cells were expanded to a final volume of 1 L in a roller bottle during a period of 8 to 10 days. The supernatant was harvested, concentrated by filtration, and dialyzed with PBS. Finally, ex-EVA1-Fc protein was purified using a protein G column in a FPLC (AKTA-GE Healthcare Life Sciences). With this strategy, the yield was between 1 and 3 mg of protein per liter. Purity was visually assessed in a 12% SDS-PAGE stained with Coomassie Brilliant Blue (Fig. 1).
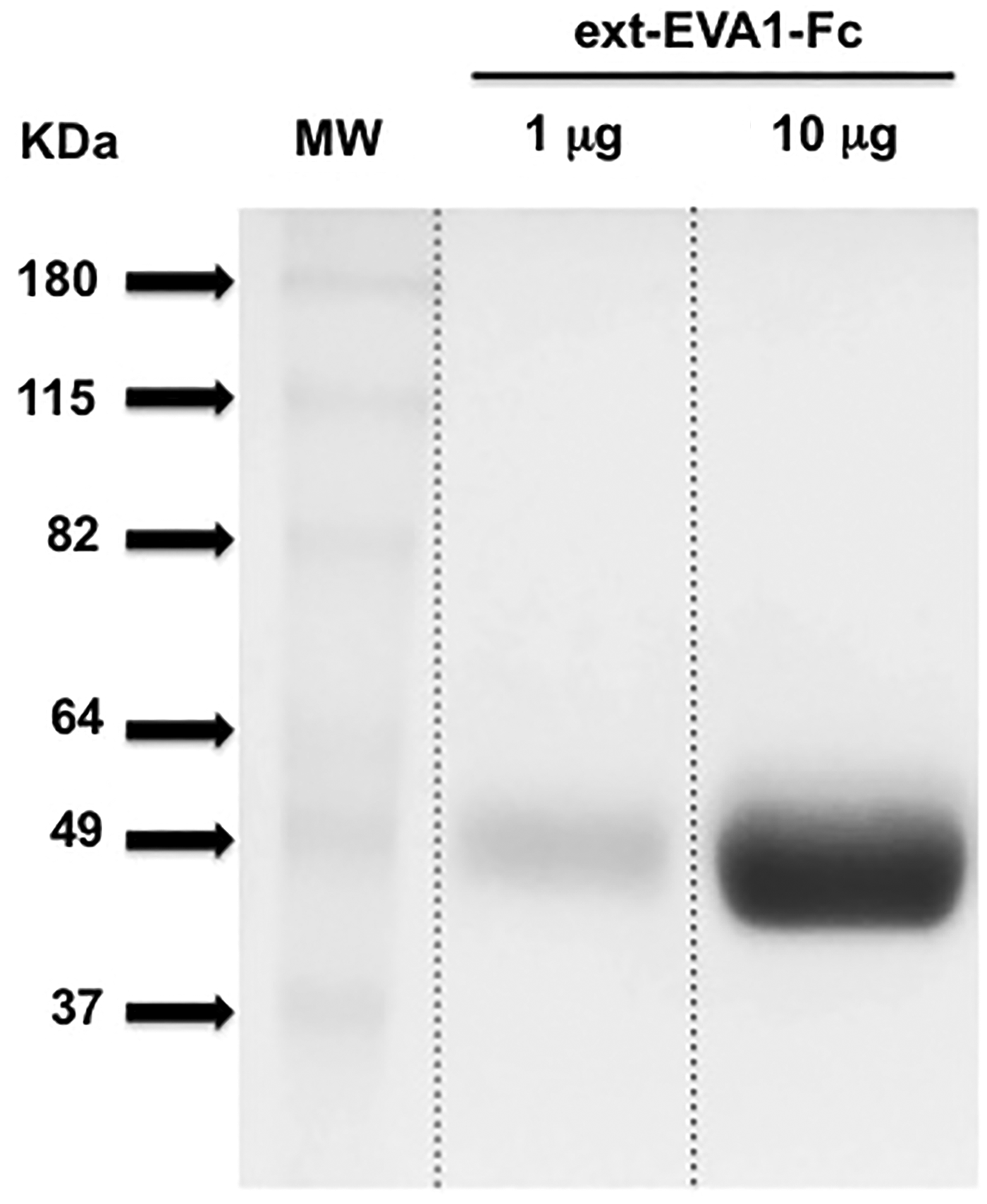
Purification of ext-EVA1-Fc protein. ext-EVA1-Fc from transduced CHO-S cell supernatant was purified with a protein G column by FPLC and purity visually assessed in a 12% polyacrylamide gel electrophoresis (1 and 10 μg of total protein) stained with Coomassie Brilliant Blue.
Establishment and reactivity of hybridoma cell lines
To perform the fusion protocol, we used splenocytes from an ex-EVA-Fc immunized mouse that produced the highest serum titer against ext-EVA1-Fc protein, as determined by ELISA. After fusion, cells were plated in eight 96-well plates (768 wells). The percentage of wells with hybridoma growth was 95%. The screening from each well was first performed by ELISA analysis of the supernatants, and the 10 highest positive wells were tested by flow cytometry on HEK-293T-YFP+ cells expressing mouse and human EVA1. We identified six positive wells, and we chose three of them to perform a single cell sorting by flow cytometry (FACSAria-BD Biosciences). For further analysis, we chose G9P3-1 clone, which secretes a monoclonal antibody (G9P3-1Mab, IgG1 kappa) reactive against mouse and human EVA1 (Fig. 2). Subsequently, two consecutive cloning of G9P3-1 were performed without detecting any change in hybridoma growth and cross-reactivity to mouse and human EVA1.
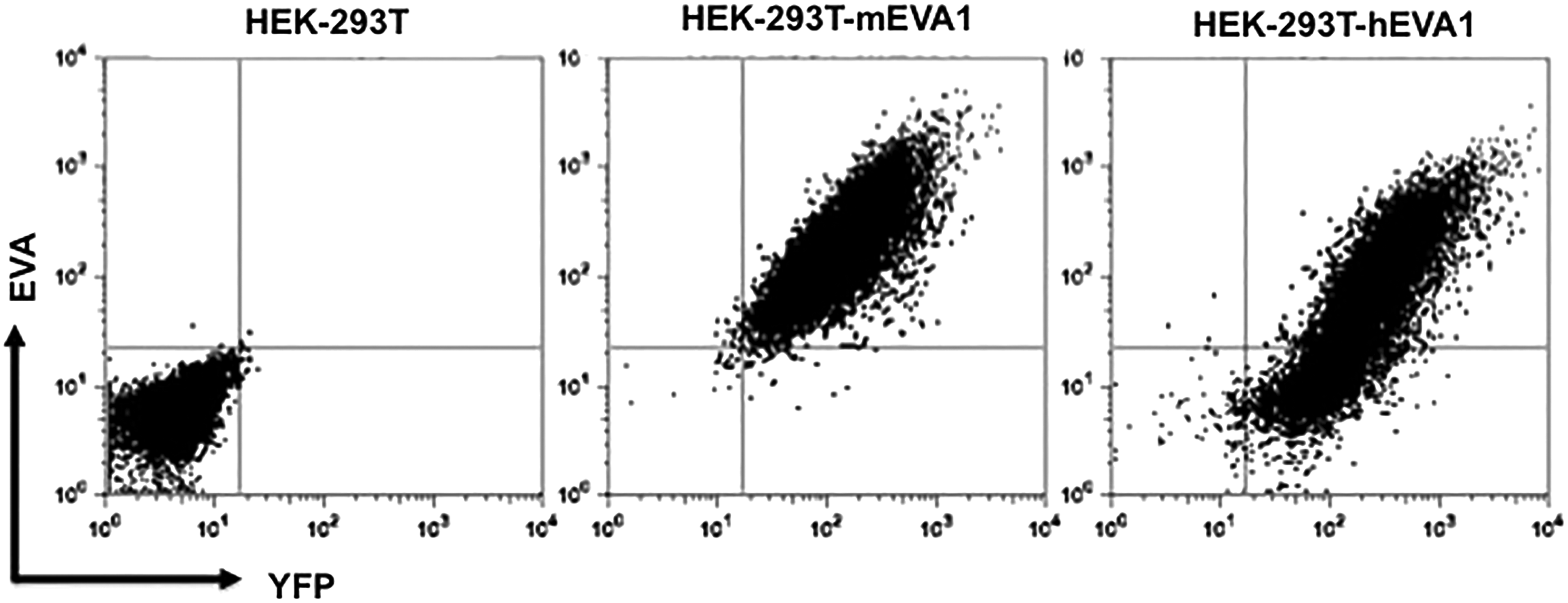
G9P3-1Mab recognizes mouse and human EVA1. HEK-293T cells transduced with retrovirus expressing mouse (HEK-293T-mEVA1) and human (HEK-293T-hEVA1) EVA1, as well as the YFP reporter, were stained with G9P3-1Mab. Flow cytometry results show that only HEK-293T cells expressing EVA1 are recognized by G9P3-1Mab.
Characterization of G9P3-1 monoclonal antibody reactivity against thymocyte and thymic epithelial subsets
Comparative analysis of Eva1 (Mpzl2) mRNA expression revealed a tightly regulated expression Eva1 at the DN3 stage of development (Fig. 3). Likewise, we found that G9P3-1Mab detected the highest expression of EVA1 on the cell surface of DN3 (CD25+CD44low) thymocytes (Fig. 4A). We used the EVA1-deficient mice (B6.Eva1ko/ko) as negative control to show the specificity of the G9P3-1 Mab to detect EVA1 expression on DN3 cells. Extended comparative analysis between two development-relevant DN3 subsets(27) DN3a (CD25+CD44lowCD28low) and DN3b (CD25+CD44lowCD28high) showed no significant differences in EVA1 protein expression, indicating that EVA1 is homogeneously, specifically, and highly expressed at the DN3 development stage.
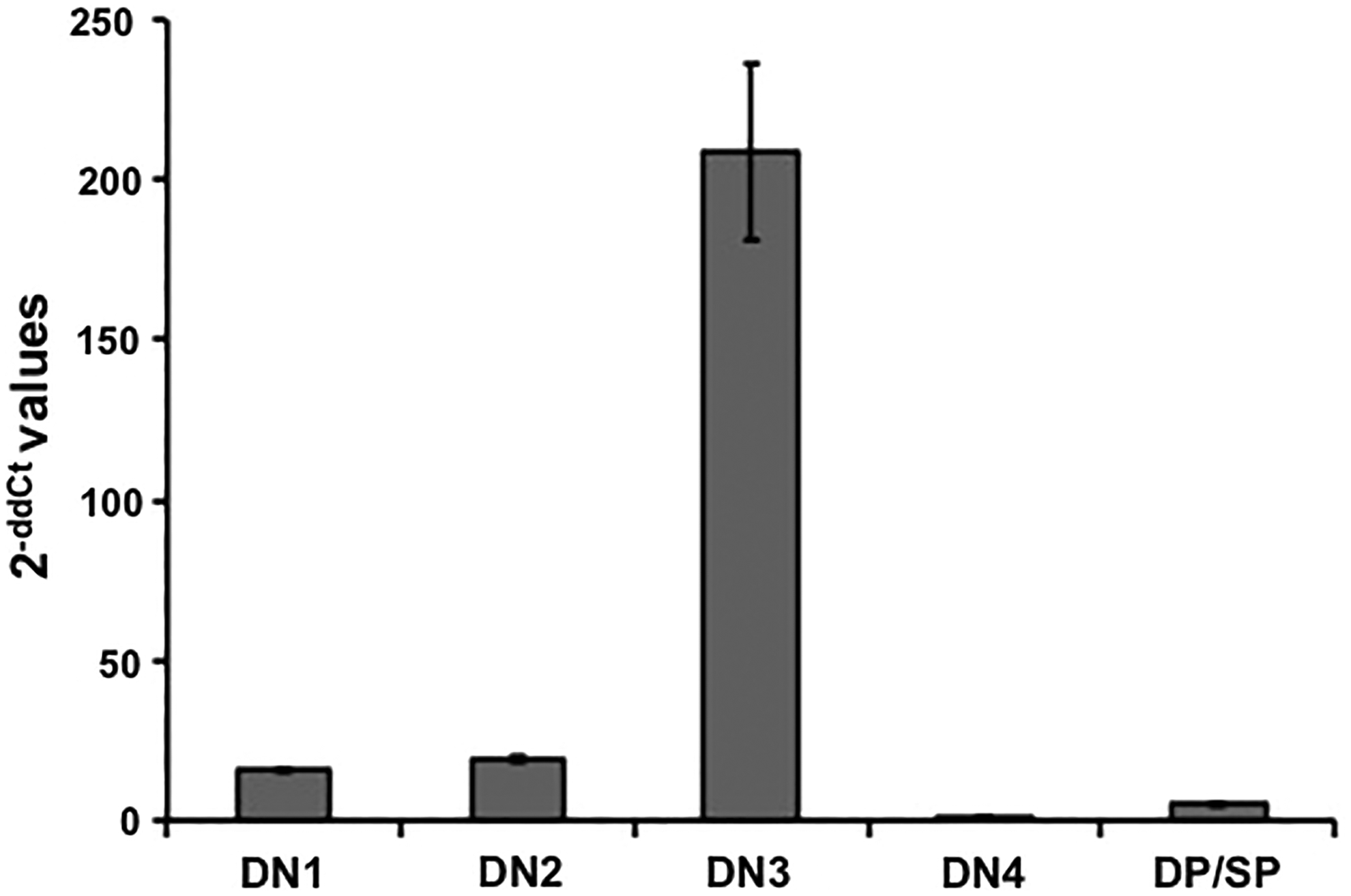
High levels of Eva1 transcripts in DN3 thymocytes. Real-time PCR results show the highest levels of Eva1 transcripts in DN3 thymocytes. Data was calculated using the 2-ddCt method,(32) which measures the relative changes in Eva1 expression. This experiment was repeated three times with similar results.
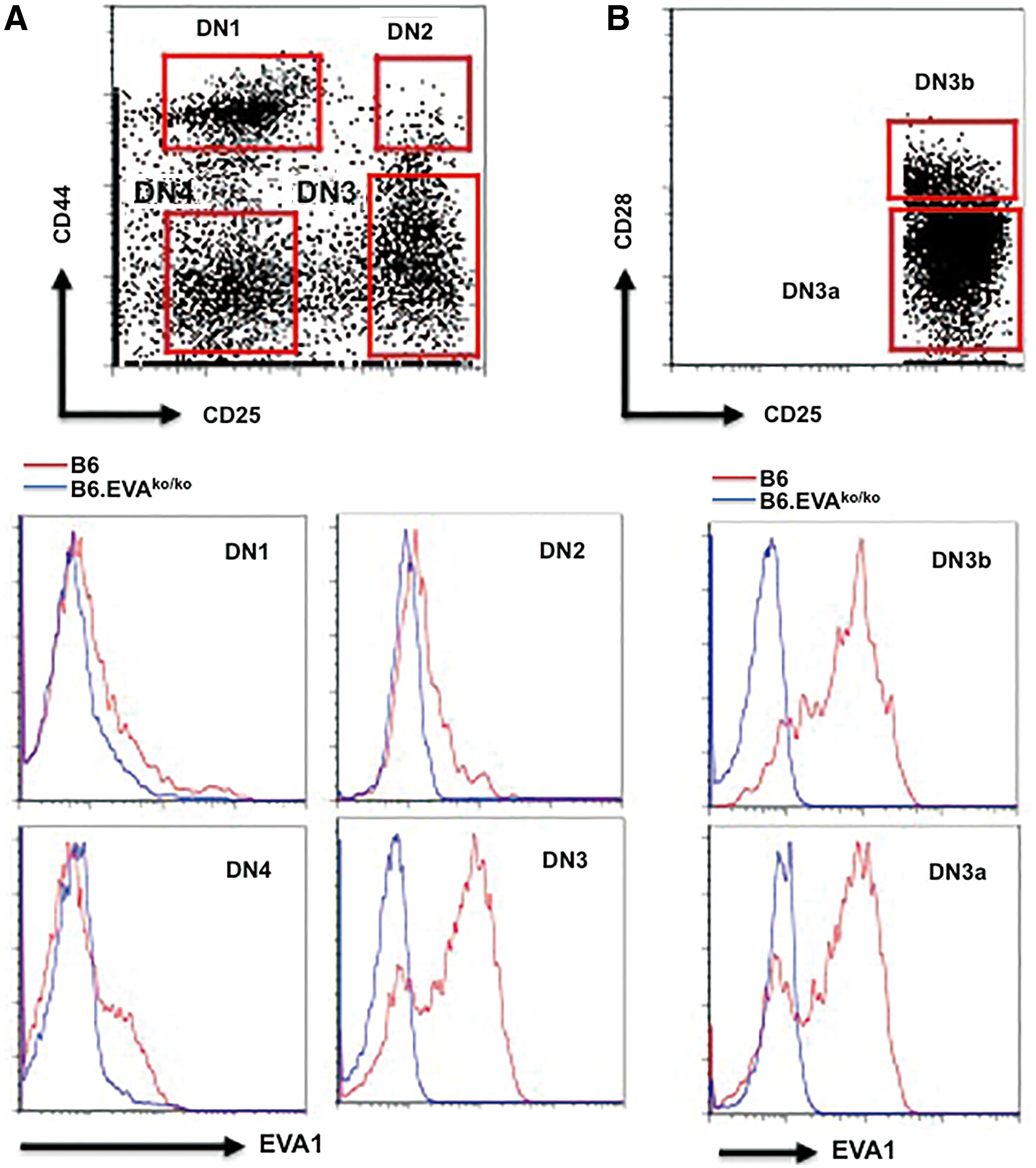
G9P3-1Mab detects expression of EVA1 on the cell surface of DN3 thymocytes. (
Additionally, we examined the expression of EVA1 on TECs, in which EVA1 was initially shown to be expressed.(19,23) Of note, G9P3-1Mab also detected EVA1 protein expression on the cell surface of cortical and medullar TECs (Fig. 5).

G9P3-1Mab detects expression of EVA1 on the cell surface of thymic epithelial cells. CD45-EpCAM+ cortical and medullar thymic epithelial cells (identified with UEA-1 FITC and Ly-51 PE) were analyzed for EVA1 expression. Both subsets (referred in the figure as medulla and cortex) express EVA1 when compared to B6.Eva1ko/ko subsets. We have not observed differences between B6 and B6.Eva1ko/ko mice in number and percentages of cortical and medullar thymic epithelial cells, and for simplicity only the UEA-1/Ly-51 profile from the B6 mouse is shown. This experiment was repeated twice with similar results.
Discussion
Although the sequence of events that contribute to thymic involution are poorly understood, disruption of thymic architecture is one of the most remarkable changes associated with this condition.(28) These changes have been associated with a decline in the expression of adhesion molecules on TECs, leading to a disruption of thymic cross-talk.(10) This cross-talk decline can also occur when changes affecting expression of thymocyte adhesion molecules are present.(15,29)
Experiments involving the adoptive transfer of Eva1 knockdown hematopoietic stem cells into thymus-involuted NOD.Scid.Il2rγ-/- immunodeficient hosts have revealed a role of EVA1 in DN3 thymocyte development and thymic regeneration.(21) In contrast, thymic cellularity and architecture appeared to be normal in the Eva1-deficient mice (PS & JCZP, unpublished results), suggesting that EVA1 may contribute to the homeostatic pathways controlling thymus regeneration. Other pro-regenerative molecules function in a similar fashion. This is the case of interleukin-22 (IL-22) that supports thymus regeneration following a discrete thymic insult. Interestingly, analysis of Il22 gene-deficient mice revealed normal thymic cellularity and thymocyte subset percentages in the absence of thymic insult.
We are presently conducting experiments to examine the potential role of EVA1 in thymus regeneration. Our preliminary data show that the protein levels of EVA1 expressed on DN3 thymocytes from a chemically involuted thymus is several orders of magnitude higher than in DN3 thymocytes from a normal thymus. We hypothesize that this increased expression of Eva1 in DN3 thymocytes contributes to counter-balance thymic involution by increasing the strength of the DN3/thymic epithelial cell cross-talk.
The homeostatic role of EVA1 may not be restricted to thymus. EVA1 has also been involved as a compensatory and upregulated adhesion molecule in testicles deficient for cell adhesion molecule-1, an adhesion molecule involved in spermatogenesis.(30)
A more common form of thymic involution is age-dependent thymic involution, one of the contributing arms to immunosenescence, a condition frequently associated with increased incidence of infections, neoplasias, and autoimmune diseases in older patients.(31) Interestingly, age-dependent thymic involution is reversible under certain circumstances, suggesting that the regenerative homeostatic pathways that are active in a post-stressed thymus could also be active in an aged thymus. Our newly developed anti-EVA1 monoclonal antibody will facilitate the study of the pathways involved in the involution and regeneration of the aged and stressed thymus.
In conclusion, our new monoclonal antibody against human and murine EVA1 is a useful reagent for studying the development and function of T cells in health and in disease. Moreover, this study illustrates the use of gene-targeted mice for the generation of monoclonal antibodies with multiple species cross-reactivity.
Footnotes
Acknowledgments
We thank Dr. Isabel Crespo, Cristina Lopez, and Laia Llinas from the cytometry facility of the Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) for technical help and Janet Astwood for text editing. This work was financially supported by a grant from the Plan Nacional (SAF2011-22458) and by the Ramón y Cajal Reintegration Fellowship from the Spanish Ministry of Science (P.S.). The JMDRC is supported by the Canadian Diabetes Association.
Author Disclosure Statement
The authors have no financial interests to disclose.