
Abstract
The complement alternative pathway (AP) is a major contributor to a broad and growing spectrum of diseases that includes age-related macular degeneration, atypical hemolytic uremic syndrome, and preeclampsia. As a result, there is much interest in the therapeutic disruption of AP activity. Properdin, the only positive regulator of the AP, is a particularly promising AP target. Several issues need to be clarified before the potential for properdin-directed therapy can be realized. In this report we use a portion of the mouse properdin protein, expressed in a bacterial system, to raise rabbit polyclonal and hamster monoclonal antibodies that block properdin-dependent pathogenesis. These antibodies, when employed with AP-dependent mouse disease models, can help evaluate the feasibility of properdin-directed therapy.
Introduction
C
There are three complement activation pathways: the classical pathway, the lectin pathway, and the alternative pathway (AP).(1,5) Each activation pathway leads to the assembly of the C3 convertases, enzymes that catalyze the cleavage of C3, on the target surface. Over the last decade the alternative pathway has emerged as a major causative agent in common and rare inflammatory diseases, including age-related macular degeneration, atypical hemolytic uremic syndrome, and preeclampsia.(6–19) As a result, the AP components have been considered as potential therapeutic targets.
Assembly of the AP convertases(5) begins with the covalent attachment of nascent C3b to a target surface followed by association of C3b with factor B (FB). The C3bB complex is then cleaved by factor D (FD) at a single FB site, forming an active but unstable (T1/2 ∼90 sec) C3 convertase, C3bBb. An additional AP protein, properdin (P), binds to C3bBb, rendering the convertase 5–10-fold more stable.(20) Properdin may also bind to certain surfaces like bacteria and apoptotic and necrotic cells and initiate the AP.(21) Because the C3bBb complex can cleave C3 in the absence of properdin,(22) the possibility of properdin-targeted therapeutics was not widely discussed until recently, when it was shown that properdin plays a critical role in AP-dependent pathogenesis in several mouse disease models.(23,24)
Properdin is not made by the liver like most complement proteins but instead is released from peripheral cells and from there accumulates in the circulation.(25) For this reason, factors such as properdin source (plasma-derived versus neutrophil-derived), biological space (intravascular versus ocular versus lungs), and therapeutic window (acute versus chronic), could all contribute to the success or failure of a properdin inhibitor. Here we describe the generation and properties of polyclonal and monoclonal anti-properdin antibodies that block AP-dependent pathogenesis in the mouse and, as such, can be used to help resolve these issues.
Materials and Methods
Animals
WT C57BL/6J were obtained from The Jackson Laboratory (Bar Harbor, ME). Properdin-deficient mice (Cfptm1Cmst, fully backcrossed to C57BL/6 background) were obtained from the Transgenic Unit of the Division of Biomedical Services at University of Leicester (United Kingdom).(26) All animal experiments were performed in compliance with federal laws and in strict accordance with the guidelines established by the Division of Comparative Medicine at Washington University. The animal protocol is subjected to annual review and approval by The Animal Studies Committee of Washington University.
Production of recombinant mouse properdin TSR5/6
The generation and purification of a His-tagged mouse TSR5/6 (mTSR5/6) has been previously described.(27) In brief, recombinant mouse properdin cDNA encoding TSRs 5 and 6 was amplified using PCR methodology and cloned into the pET28a+ expression vector (EMD/Millipore, Billerica, MA). The resulting plasmids were transformed into E. coli strain BL21(DE3) RIL codon plus (Agilent Technologies, Santa Clara, CA). Cell cultures were grown and harvested following IPTG induction. Inclusion bodies were isolated and denatured in guanidine/TCEP buffer, protein refolded at 100 μg/mL, and aggregates removed by centrifugation. Refolded protein was concentrated and its identity confirmed and level of purity determined by gel electrophoresis/Coomassie blue stain and Western blot employing anti-mouse properdin polyclonal antibody.
Generation of rabbit anti-mouse properdin polyclonal antibodies
Purified mTSR5/6 was used to raise rabbit antibody (Harlan Laboratories, Madison, WI). Antibody was purified from the immune serum by protein G chromatography.
Generation of hamster anti-mouse properdin monoclonal antibodies
His-tagged mTSR 5/6 (above) was used to immunize Armenian hamsters. Spleens from immunized animals were fused with Sp20 myeloma cells by standard protocol established by the Hybridoma Center at Washington University.(28) Clones were selected by an ELISA-based assay. Several reactive clones were selected for further subcloning and characterization. To obtain purified MAbs, the hybridoma cell lines were grown until 95% cell death to produce hybridoma exhausted supernatants. MAbs were purified from exhausted supernatants on a protein-G column. Properdin specificity was confirmed by a Western blot analysis that compared antibody reactivity to normal mouse serum versus serum derived from properdin-deficient mice.(27) Clones H4 and E12 were selected for the studies presented herein.
Assessment of mouse alternative pathway activity in vivo
An ELISA was employed to evaluate the AP activity of plasma derived from antibody-treated animals.(29,30) Mice were injected i.p. with 1.5 mg of H4 or E12 MAbs. Freshly prepared plasma samples were collected at various time points (1–14 days), diluted (1:20 in Mg2+-EGTA buffer(31)), applied to LPS-coated ELISA plates (#3855; Thermo Fisher Scientific, Waltham, MA) (2 μg/well) (cat #L2762; Sigma-Aldrich, St. Louis, MO), and incubated at 37°C for 1 h. After washing, goat anti-mouse C3 Ab (1:4000 dilution, cat #55463; MP Biomedicals, Santa Ana, CA) was added followed by HRP-conjugated anti-goat IgG (1:2000 dilution, cat #705-035-147; Jackson ImmunoResearch Laboratories, West Grove, PA), after which substrate reagent (cat #DY999; R&D Systems, Minneapolis MN) was added for 10 min. The reaction was stopped with 1 M H2SO4 and the OD of samples were measured at 450 nm (SpectraMax Plus 384, Molecular Devices, Sunnyvale, CA).
Elastase-induced abdominal aortic aneurysm mouse disease model
Abdominal aortic aneurysm (AAA) was induced in 8- to 12-week-old male mice as previously described.(24,27,32,33) Briefly, mice were anesthetized with 55–60 mg/kg intraperitoneal sodium pentobarbital. A laparotomy was performed under sterile conditions. The abdominal aorta was isolated and the pre-perfused AD was measured with a calibrated ocular grid. Temporary 7-0 silk ligatures were placed around the proximal and distal aorta to interrupt proximal flow. An aortotomy was created at the inferior ligature using the tip of a 30-gauge needle, and the aortic lumen was perfused for 5 min at 100 mm Hg with a solution containing 0.145 U/mL type 1 porcine pancreatic elastase (Sigma-Aldrich). After removal of the catheter, the aortotomy was repaired without constriction of the lumen to restore the flow. At day 14, a second laparotomy was performed and the perfused segment of the abdominal aorta was re-exposed and unblinded quantitative digital readout of AD was obtained in situ by two individual observers prior to euthanasia and tissue procurement. AD was assessed on day 14 unless otherwise indicated. AAA is defined as an increase in AD of greater than 100% over pre-perfused diameter. For the studies here, mice were randomly assigned to receive an i.p. injection of 1.5 mg of rabbit polyclonal anti-mTSR5/6 antibodies or non-immune rabbit IgG, H4, E12, or Armenian hamster control IgG (cat#IR-AHT-GF, Innovative Research, Novi, MI) at 1 h prior to elastase perfusion, immediately after and 24 h after surgery (for a total of three doses).
Statistical analysis
Comparisons between multiple groups (≥3) were performed by one-way ANOVA. Equality of variance assumption was tested and Bonferroni's correction for multiple comparisons was performed. The sample size (number of animals per genotype/treatment) chosen is based on means and variances in similar experiments in this mouse model of AAA for detection of differences between experimental groups at an alpha level of 0.05 and a statistical power of 0.80, assuming a two-sided test. F test was used to compare variances within each group of data, and the difference in variances was found to be not significant between groups.
Results
Rabbit polyclonal antibody produced with mouse TSR5/6 blocks AP-dependent pathogenesis in vivo
Properdin is a polymeric glycoprotein composed of identical 53KD subunits associated head-to-tail in dimeric, trimeric, and tetrameric assemblies.(34) The properdin subunit is a single amino acid chain composed of seven thrombospondin type 1 repeats (TSRs) numbered in order from the N terminus TSR0 followed by TSR1–6.(35,36) Several observations mark TSRs 5 and 6 as essential to properdin function: Higgins and colleagues have shown that properdin constructs lacking TSR5 were deficient in C3b-binding.(37) Fredrikson and associates showed that individuals bearing the TSR6 substitution Y387D were at risk for fulminant meningococcal disease and generated properdin multimers that lacked in vitro activity.(38) Perdikoulis and associates showed that polyclonal antibodies raised against human TSR5 were capable of blocking properdin activity in vitro.(39) From these findings we reasoned that antibodies raised against an antigen that encompasses mouse TSR5 plus mouse TSR6 region (mTSR5/6) would likely block properdin function. To that end we employed a bacterial expression system to produce mouse TSR5/6 tagged with 6xHis residues at its amino terminus (mTSR5/6).(27) Purified mTSR5/6 was then used to raise rabbit polyclonal antibody (see Methods section), and IgG was purified from immune serum by protein A chromatography. The resulting polyclonal antibody (anti-mTSR5/6) recognized both mouse and human properdin (Fig. 1).
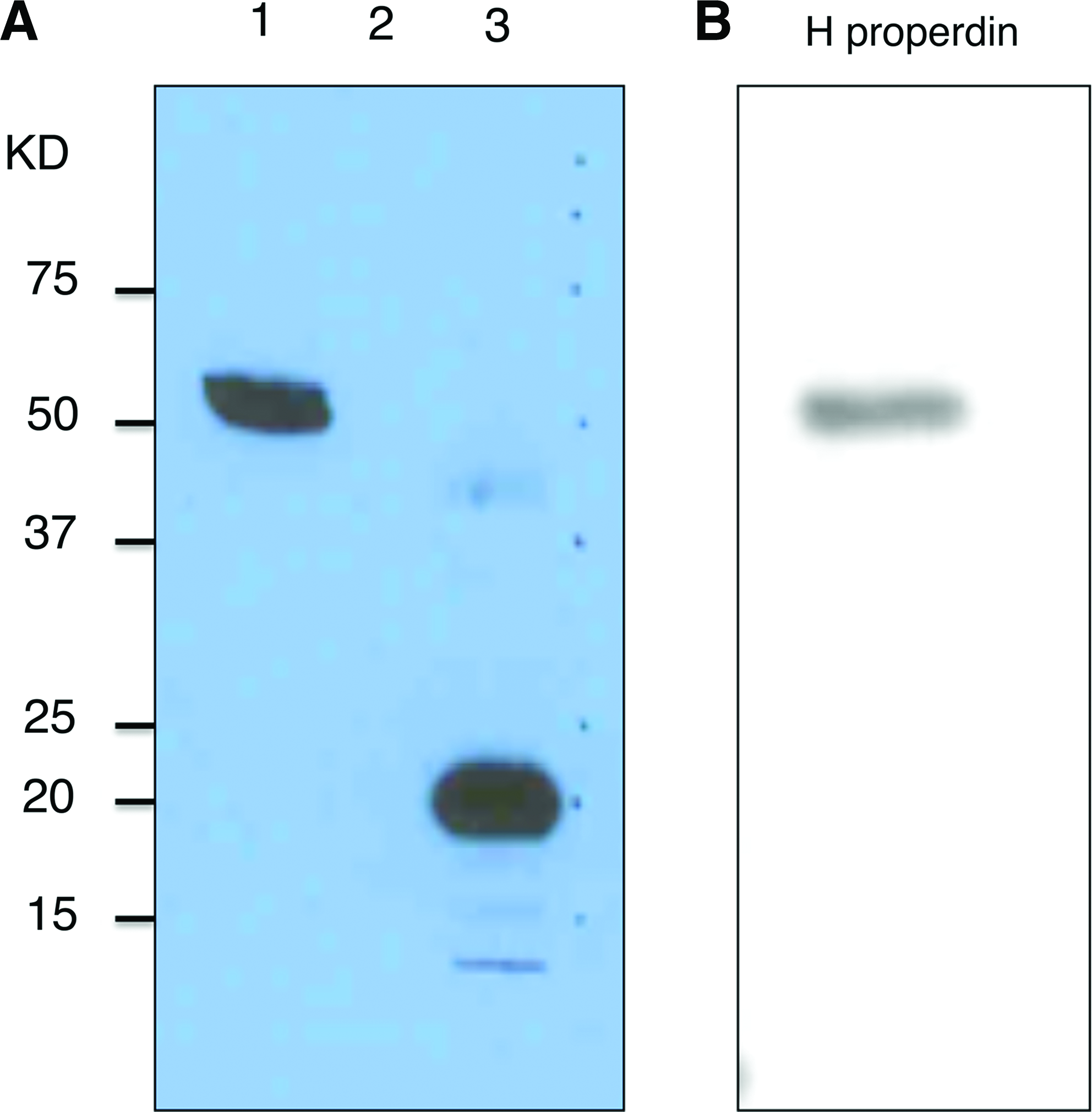
Western blot analysis of rabbit anti-mTSR5/6 polyclonal antibody. (
To test the activity of rabbit anti-mTSR5/6 antibodies in vivo, we used the elastase-induced abdominal aortic aneurysm (AAA) mouse model(40) that recapitulates many of the key immune and inflammatory features of the human disease. We had previously established that aneurysm development requires complement activity and that complement depletion prior to or up to 24 h following elastase perfusion largely blocks aneurysm formation.(33) Moreover, a rabbit polyclonal antibody raised against the full-length purified mouse properdin significantly attenuates aneurysm formation.(24) The rabbit anti-mTSR5/6 antibodies were found to block AAA to the same extent as we had observed with the function-blocking polyclonal antibody raised against full-length mouse properdin (Fig. 2). Similar results were obtained with polyclonal rabbit anti-mTSR5/6 antibodies prepared from a second immunized rabbit (Fig. 2). These results clearly demonstrate the utility of bacterially expressed properdin fragments in the generation and/or selection of properdin antagonists and the potential of anti-mTSR5/6 antibodies for assessing the importance of properdin in other C-dependent pathologic conditions.
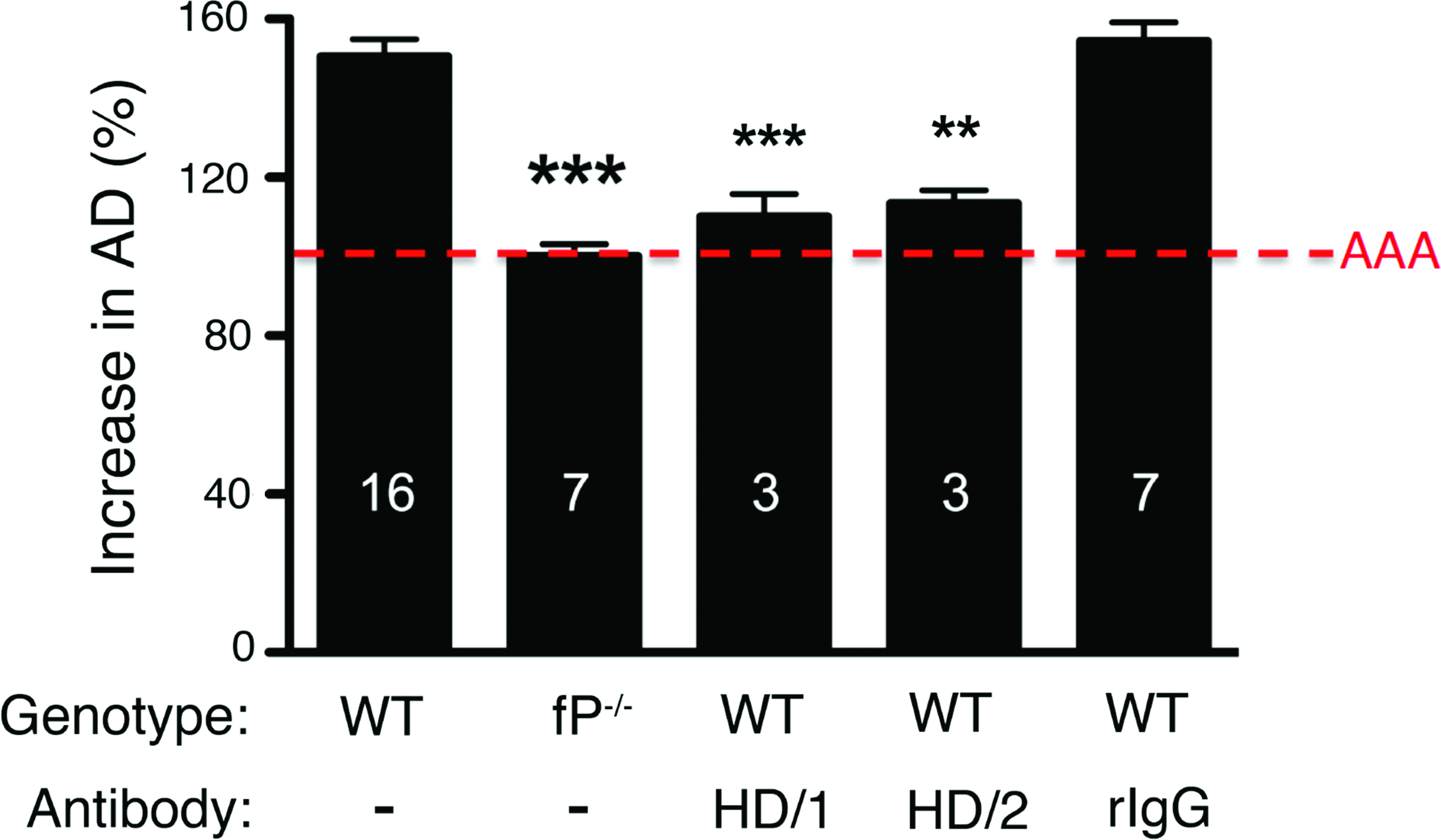
Rabbit anti-mTSR5/6 polyclonal antibody attenuates AAA development. WT and properdin-deficient (fP-/-) mice were perfused on day 0 with elastase (detailed in Methods section). WT animals were injected i.p. with 1.5 mg of the indicated anti-mTSR5/6 rabbit polyclonal antibody (HD/1 or HD/2, Abs derived from different immunized animals) or normal rabbit IgG (rIgG) as control 1 h prior to and immediately after surgery and on 24 h post-surgery. On day 14 following elastase perfusion, the aortic diameter (AD) was reassessed and increase in AD was expressed in percentage (%). AAA is defined as an increase in AD of >100% over the pre-perfused diameter (red dotted line). Values represent mean±SEM; the number of animals is indicated for each genotype and treatment. **p<0.01, ***p<0.001 compared with untreated WT.
Hamster monoclonal antibodies raised against mouse TSR 5/6 block AP-dependent pathogenesis in vivo
Given the successful generation of function-blocking polyclonal antibody raised against mTSR5/6, we next used this antigen to generate hamster monoclonal antibodies.(28) Hamster was chosen because it provides MAbs that are non-immunogenic in mice. Hybridoma supernatants were characterized by an ELISA-based assay and the most reactive clones were selected for subcloning and characterization by Western blot. Two MAb lines (H4 and E12), derived with different animals, were purified using a protein-G affinity column and used in the studies here.
We first evaluated the effects of H4 and E12 on alternative pathway activity in vivo. To that end, mice were injected i.p. with 1.5 mg of purified H4 or E12 and plasma was collected at various intervals and assayed for LPS-dependent AP activity.(29,30) Naïve WT served as positive controls and properdin-deficient mice(26) served as negative controls. Both H4 and E12 abrogated alternative pathway activity and full inhibitory activity lasted at least 14 days (Fig. 3).

Hamster anti-mTSR5/6 monoclonal antibody blocks the complement AP in vivo. 1.5 mg of the indicated MAb was injected i.p. into mice (two animals per time point). Plasma was collected at the indicated time point and assayed for AP activity as indicated by LPS-dependent surface deposition of C3 fragments (see Methods). Properdin-deficient (fP-/-) plasma served as negative control.
We next assessed whether the H4 and E12 MAbs block elastase-induced AAA development. H4 and E12, but not the Armenian Hamster control IgG, significantly attenuated AAA formation (Fig. 4). Taken altogether these results show that anti-mTSR5/6 MAbs are valuable reagents that can be used to dissect the contribution of properdin and of the alternative pathway in complement-dependent disease processes.
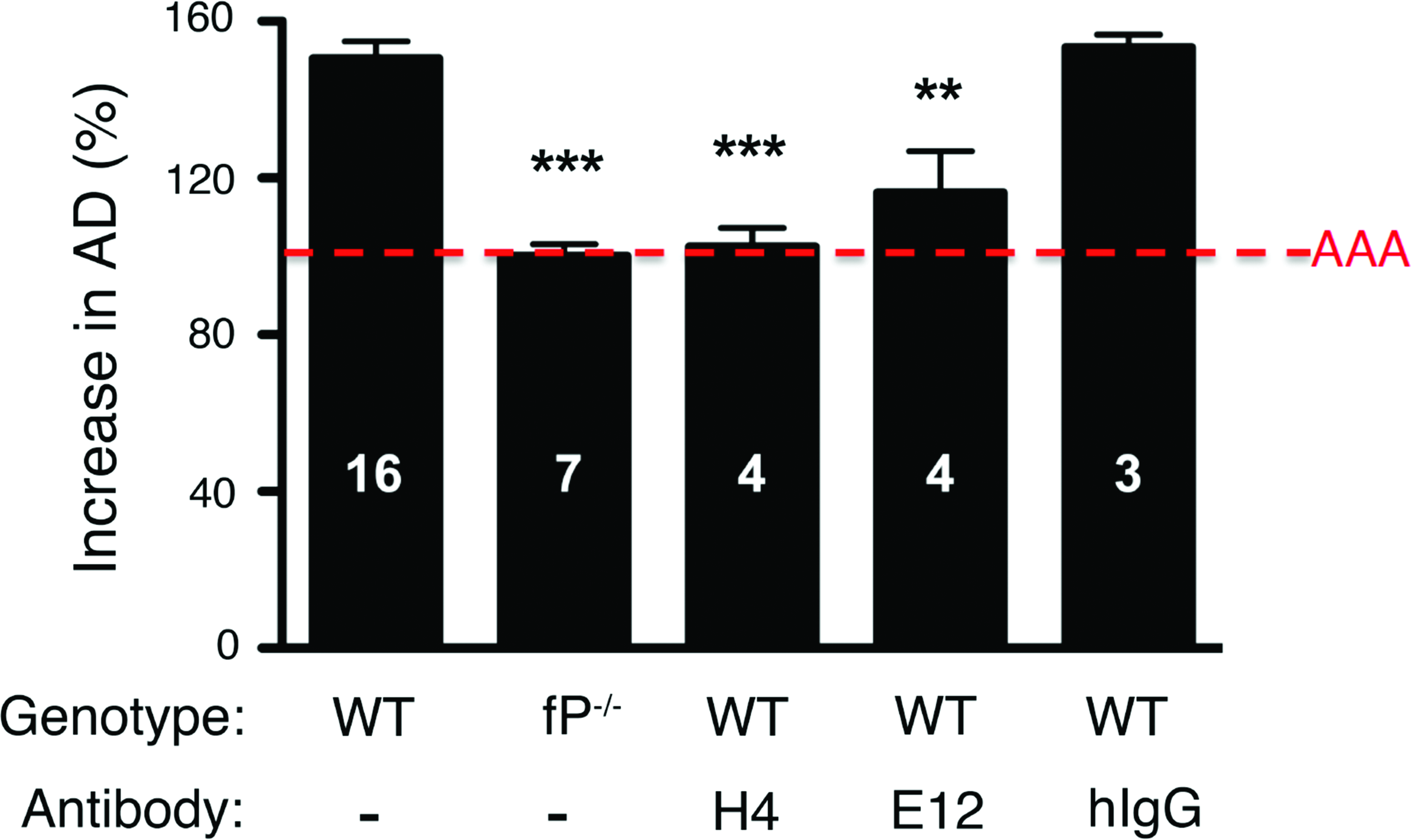
Hamster anti-m TSR5/6 monoclonal antibody attenuates AAA development. WT and properdin-deficient (fP-/-) mice were perfused on day 0 with elastase. WT animals were injected i.p. with 1.5 mg of the indicated anti-TSR5/6 monoclonal antibody (H4 or E12) or Armenian hamster isotype control (hIgG) 1 h prior to and immediately after surgery and 24 h post-surgery. On day 14 following elastase perfusion, the aortic diameter (AD) was reassessed and increase in AD was expressed in percentage (%). AAA is defined as an increase in AD of >100% over the pre-perfused diameter (red dotted line). Values represent mean±SEM; the number of animals is indicated for each genotype and treatment. **p<0.01, ***p<0.001 compared with untreated WT.
Discussion
The complement alternative pathway plays a critical role in numerous autoimmune and inflammatory diseases.(2) Among the AP components, properdin is a particularly attractive therapeutic target given that it is unique to the AP, is present at a relatively low concentration in the plasma (10-fold lower than factor B, 50-fold lower than C3(41)), and its stoichiometric mode of action may make it particularly amenable to inhibition. Moreover, properdin-deficient individuals (nearly all males, as the properdin gene is located on the X chromosome(42)) are generally healthy except for a marked susceptibility to fulminant meningococcal disease, a condition that usually can be addressed by prophylactic vaccination.(43)
This study was initiated to produce hamster MAbs that block mouse properdin activity in vivo. Rather than using native mouse properdin as antigen, we expressed in a bacterial system a region of mouse properdin previously implicated in AP function. We first determined that mTSR5/6 could be used to raise function-blocking rabbit anti-mouse properdin polyclonal antibody. We next generated from two different immunized hamsters, hybridoma lines that produced function-blocking monoclonal antibodies. Recent EM studies indicate that the amino- and carboxy-terminal regions of the properdin monomers associate together in globular regions that bind both C3b and Bb, thus accounting for the properdin stabilizing effects.(44) The antibodies we describe here, raised against the carboxy-terminal domains, likely interfere with one or more of these interactions.
Properdin is not synthesized in the liver like most complement proteins. Instead, it is secreted by T cells, monocytes, mast cells, and granulocytes and is stored in neutrophil granules and released at sites of inflammation.(25) AP activity can be promoted by properdin released locally or by properdin that has accumulated in the circulation. In the case of elastase-induced AAA, for example, pathogenesis can be restored in properdin-deficient animals by systemic treatment with WT plasma and can also be partially recovered by the administration of WT neutrophils.(24) Thus, therapeutic inhibition of properdin activity may need to be addressed at systemic and/or local level(s). In addition, the consequences of properdin deficiency must be fully investigated before the general use of properdin-targeted therapy can be considered.(45) The hamster anti-mTSR5/6 MAbs we describe here are useful tools that, when employed with AP-dependent animal models, can help resolve these and other related issues.
Footnotes
Acknowledgments
The authors would like to thank Dr. Kathleen Sheehan, Misha Hart, and Paul Schjetnan for the production of Armenian hamster hybridoma cell lines; Marilyn Leung for help in hybridoma screening; Richard Hauhart for antibody purification; and Madonna Bogacki for assistance in preparing the manuscript. This work was supported by an NIH grant (no. AI051436 to D.E.H.). Experimental support was also partially provided by the Hybridoma Center and the Protein Production and Purification Facility of the Rheumatic Diseases Core Center (NIH P30AR048335). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Author Disclosure Statement
Dennis Hourcade is a consultant for Alexion Pharmaceuticals (Cheshire, CT).