
Abstract
Orf is caused by the orf virus (ORFV) and is a non-systemic, widespread disease afflicting sheep, goats, wild ruminants, and humans. Recent outbreaks in sheep and goats in Jilin and other northern Chinese provinces raise concerns about orf control in China. Thirty-five hybridoma clones were constructed from splenocytes of BALB/c mice immunized with natural orf virus protein. These hybridomas were used to produce antibodies targeting ORFV proteins. Immunological characterization of these monoclonal antibodies (MAb) showed that the 5F2D8 hybridoma line produced MAb that can recognize the 100, 70, and 20 kDa bands from total viral lysate. This hybridoma was further characterized by immunoprecipitation and peptide sequencing. The results indicate that 5F2D8 specifically recognizes orf virus encoded protein ORFV086, a late expression virion core protein that plays important roles in progeny virus particle assembly, morphogenesis, and maturity. Further experiments demonstrate that this MAb did not react with other viral proteins of ORFV orthopoxviruses, but reacted strongly to different field isolates of orf viruses from China. Additionally, this anti-ORFV086 MAb possesses ORFV neutralizing capability. Sequence alignments and phylogenetic analysis determined that ORFV086 of NA1/11, clustered together with NZ2 and IA82, is highly conserved and has structural similarities with the Vaccinia virus core protein P4a. As such, this MAb has great potential as a diagnostic tool for orf viruses, in the further exploration of orf pathogenesis, and in disease control and prevention.
Introduction
O
ORFV is a Parapoxvirus of the subfamily Chordopoxvirinae and the family Poxviridae.(7) Bovine papular stomatitis virus and pseudocowpox virus are additional members of this genus.(7) The complete genome of the orf virus is 138 kb, contains 64% GC, and includes 132 putative genes.(8) While a number of immunogenic proteins have been identified in convalescent sera taken from infected sheep,(9) the mechanisms of host immunity to orf infection are not well understood and protective antigens need to be identified. In several provinces in northern and eastern China, recent outbreaks of orf have afflicted sheep and goat stocks.(10,B11) Thus far, identification and characterization of the strain or strains responsible for these outbreaks of orf (NA1/11) have been limited and investigations into the prevalence of NA1/11 in China have been scarce.(12) Therefore, MAbs against ORFV-Jilin, for use as diagnostic tools, are urgently required.
To address this lack of information about orf in China, and to identify more immunogenic proteins, we have set about generating MAb capable of recognizing NA1/11. Briefly, purified viral proteins were used in the production and cloning of mouse hybridoma line that produced monoclonal antibodies against ORFV. Subsequently, the MAb was purified and analyzed, indicating that it recognizes the ORFV086 protein and reacts strongly. The predicted size of ORFV086 protein is 100.05 kDa. The ORFV086 gene encodes a structural protein that is expressed on the intracellular, mature virus core and shares structural similarities with the vaccinia virus (VV) precursor core protein P4a and other poxvirus homologues.(13–15) For vaccinia virus, proteolytic maturation of the most abundant major structural protein p4a leads to release of the 62, 23, and 9 kDa products, and is essential for the formation of mature infectious VV progeny.(B14,B15) Furthermore, the p4a major core protein forms a stable complex with a vaccinia virus 39 kDa protein(16) encoded by the VV A4L gene early in morphogenesis, which shares similarities with the ORFV080 gene. As a diagnostic core reagent, 5F2D8 may prove to be highly valuable for further examinations regarding ORFV086 protein proteolysis, viral assembly, the processes of orf pathogenicity, and orf disease control.
Materials and Methods
Cells and virus
Primary ovine fetal turbinate (OFTu) cells (Southern Medical University, Guangzhou, Guangdong, China) were used in the experiments and were cultured in minimal essential medium (MEM; Hyclone, Logan, UT) containing 2 mM 1-glutamine, 100 μg/mL streptomycin, 50 μg/mL gentamicin, and 100 U/mL penicillin. The media was supplemented with 10% FBS (Gibco, Grand Island, NY). The ORFV NA1/11 strain used in this study was isolated from a sheep in the northeastern Chinese province of Jilin.(10) Different ORFV strains, isolated recently from the Fujian(11) and Henan Provinces(17) of southern and central China, were used in the cross-reactivity study (Table 1), along with vaccinia virus,(12) fowl poxvirus,(12) and goatpoxvirus.(18)
The reactivity of 5F2D8 with different proteins of ORFV was determined by ELISA. Positive reactivity is indicated by a plus (+) and negative reactivity by a dash (–).
Ethics statement
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at South China Agricultural University (certification no. CNAS BL0011).
Purification of ORFV-NA1/11
Ovine fetal turbinate cells were grown in 10 T150 tissue culture flasks and, when 90% confluent, were infected with NA1/11 (5 MOI). When cytopathic effect was evident (in 3–5 days) and the majority of cells displayed a rounded morphology, the media was centrifuged (1500 rpm, 4°C, 10 min). The supernatant was subjected to sucrose gradient ultracentrifugation(19,20) to separate the viral particles from other matter. The viral particles were removed and heat-inactivated for 90 min at 96°C, sonicated, and stored at −80°C. A protein assay reagent (Bio-Rad, Hercules, CA) was utilized to determine the protein concentration of the purified virus.
Immunization of mice with purified viral protein and establishment of hybridoma lines
Ten eight-week-old female BALB/c mice were immunized via intraperitoneal (i.p.) injection with 50 μg of viral protein and bentonite (Thermo Fisher Scientific, Waltham, MA). Boosting of mice started 4 weeks after initial immunization, and consisted of i.p. injection of 50 μg viral proteins four times per week in 2-week intervals. Splenocytes were harvested from the mice and used to establish hybridoma lines as previously described.(12) Briefly, splenocytes were washed twice with PBS, and fused with SP2/0 myeloma cells (5:1 ratio), using polyethylene glycol 3500 (Roche, Mannheim, Germany) as a fusogen. The fused cells were re-suspended in RPMI 1640 medium (Hyclone, Logan, UT) supplemented with 20% FBS, 10 U/mL IL-6 (Roche), OPI media supplement (Sigma-Aldrich, St Louis, MO), and HAT media supplement (Sigma-Aldrich). They were then plated into 96-well tissue culture plates (1.2×105 cells per well) in a volume of 200 μL. After incubation for 7 to 10 days (37°C, 5.0% CO2), the culture medium in each well was analyzed by indirect ELISA to detect the presence of MAb against viral protein. Several hybridoma lines were validated, and 5F2D8 was selected for further investigation after it was identified by mass spectrometry and bioinformatics analysis. While 5F2D8 was specifically chosen for this study, all of the other hybridoma lines were further characterized. Hybridoma line 5F2D8 was subjected to limiting dilution three times before being separately amplified in mice. As previously described,(12) each of five pristine-treated mice received 8×105 cells in 0.5 mL of PBS by intraperitoneal injection. The mice were euthanized under terminal halothane anesthesia after 10 days and the ascitic fluid was collected. The fluid from the mouse ascites was centrifuged at 12,000 rpm (4°C, 10 min) to separate cellular debris and stored at −20°C. The MAb was purified using Protein G Sepharose 4 Fast Flow media (GE Healthcare, Piscataway, NJ), following the manufacturer's instructions.
Enzyme-linked immunosorbent assay
Detection of MAb was performed by ELISA, as previously described.(12) Briefly, ELISA plates were coated with purified viral protein (100 μL, 1 μg/mL, 4°C, overnight), blocked with 1% BSA (2 h, 4°C), and incubated with 100 μL of hybridoma cultured medium (1 h, 37°C). The wells were washed with PBS and incubated 30 min with HRP-conjugated goat anti-mouse antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). The plates were then washed 4 times with PBS and incubated 10 min with ABTS substrate (Roche). The OD405 of the solutions was then measured.
Immunoprecipitation, Western blot, and microsequence analyses
For immunoprecipitation, purified viral proteins were incubated with purified MAb 5F2D8 (4°C) and allowed to react. After 3 h, 50 μL of protein G agarose beads (Millipore, Bedford, MA) were added and allowed to incubate (4°C) overnight. The following day, the samples were washed with lysis buffer three times. SDS-PAGE (10%) was used to resolve the immunoprecipitated proteins. The separated proteins were stained with GelCode Blue. For Western blots, precipitates were separated via 12% SDS-PAGE and transferred to PVDF membranes (Bio-Rad). Blots were probed with purified 5F2D8, washed, and incubated with goat anti-mouse HRP-conjugated IgG antibody (Santa Cruz Biotechnology). The membranes were washed and developed using an enhanced chemiluminescent substrate (ECL, Thermo Scientific). For peptide sequencing, immunoprecipitated proteins were excised from the gels, digested with trypsin, and separated. The peptides were identified via MADLI-TOF-TOF mass spectrometry (
Immunofluorescence
Immunofluorescent examination was performed as previously described.(12) Briefly, OFTu cells were seeded and grown on glass cover slips. These cells were infected with ORFV-Jilin (5 MOI). At 0, 3, 5, 8, 10, 12 and 24 h post-infection, the cells were fixed with 4% formaldehyde (Sigma-Aldrich), and permeabilized with 0.25% Triton X-100 (10 min, RT). After blocking with BSA in PBS, the cells were incubated with MAb 5F2D8 for 1 h at RT. Unbound antibody was washed away and the samples were incubated with anti-mouse IgG Alexa Fluor 488 (Molecular Probes, Carlsbad, CA) for 1 h at RT. Finally, the samples were stained with DAPI (10 min) (Molecular Probes). The samples were examined under an inverted fluorescence microscope (Leica, Wetzlar, Germany).
Virus neutralization assays
The ability of MAb 5F2D8 to neutralize the infectivity of ORFV-Jilin was examined by using a modified fluorescent focus neutralization assay,(B12,21) as described previously.(12) Briefly, 50 μL of 4 ten-fold dilutions of purified 5F2D8, normal mouse serum in MEM supplemented with 5% FBS or simply MEM supplemented with 5% FBS were added to individual wells of a 96-well plate. Virus was added to each well (50 μL, 2000 TCID50/mL) and incubated for 1 h at 37°C. The contents of each well were transferred to separate wells of a 96-well plate containing confluent monolayers of OFTu cells. After 24 h, the monolayers were fixed with 4% formaldehyde (1 h, RT), and the infected cells were detected based on reactivity with 5F2D8 using the Abcam immunofluorescence protocol (
Immunohistochemistry
Skin samples were obtained from a diseased sheep and examined for morphological changes consistent with ORFV infection. Infected tissues were cut into 4 μm thick sections and mounted on slides pre-treated with silicon. Endogenous peroxidase activity was quenched with 3% H2O2 (RT, 15 min). The samples were washed four times, at 5-min intervals, with PBS (pH 7.2) and blocked with 5% BSA in PBS (15 min, RT). The samples were then incubated overnight (4°C) either with diluted, purified 5F2D8 (1:1000 in PBS, 1% BSA) or with diluted normal mouse serum. Following overnight incubation, the sections were washed three times with PBS and incubated (4 h, RT) with a diluted HRP-conjugated rabbit anti-mouse IgG antibody (1:2000, in PBS, 1% BSA; Cell Signaling Technology, Boston, MA). A final wash was performed by rinsing three times with PBS. The samples were treated with a working solution of DAB (3,3′-diaminobenzidine-tetrahydrochloride) from a substrate kit for peroxidase (Promega, Madison, WI), according to the manufacturer's instructions. Positive reactions were then visualized. Afterward, slides were counterstained with hematoxylin, washed in PBS for 10 min, and mounted with GVA medium (Promega).
Polymerase chain reaction and sequencing
Polymerase chain reaction (PCR) and sequencing were performed as previously described.(10) Briefly, DNA was extracted from skin lesions, cultures of infected cells, and purified virions. Based on the OV-IA82 genomic sequence,(8,22) two primers were designed to amplify the entire open reading frame of ORFV086, using NA1/11 genomic DNA as template. PCR was carried out in a GeneAmp PCR 2400 thermocycler (PerkinElmer, Shelton, CT) using a 50 μL reaction volume containing 10 μL of 5x PCR buffer (10 mM Tris–HCl and 50 mM KCl), 2 μL of DNA template, 200 μM dATP, dTTP, dCTP, dGTP, 0.4 μM of each primer, 25 μM MgCl2, and 0.5 μL of Taq polymerase (Promega). Amplification was executed by 30 cycles of denaturation at 94°C for 1 min, annealing at 58°C for 30 s, and extension at 72°C for 1 min, 30 s. PCR was ended after 10 min at 72°C. The amplified DNA products were resolved by electrophoresis in 1% agarose gel and analyzed with an IS-1000 Digital Imaging System (Alpha Innotech, San Leandro, CA). The amplicons were ligated into the TA cloning Vector (Invitrogen, Carlsbad, CA), following the manufacturer's instructions. Automated nucleotide sequencing was performed (DNA Sequencer 373A, Applied Biosystems, Norfolk, CT) in both orientations using sequencing primers T7-promotor and M13-reverse −24. The sequences were edited using Sequencer, v3.0 (Gene Codes, Ann Arbor, MI). The sequences were edited, aligned, and deposited in GenBank (accession no. JQ729675.1).
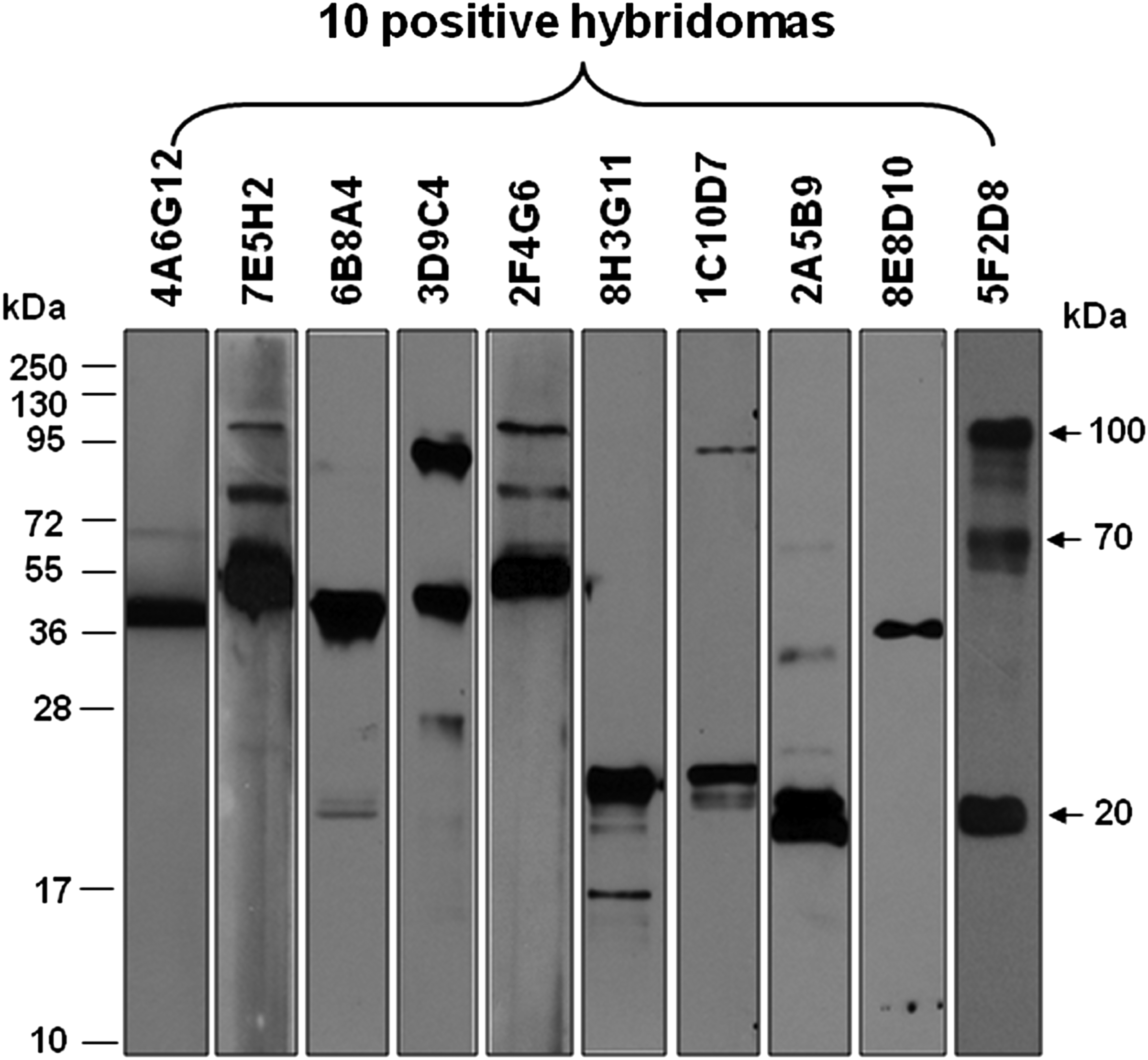
Western blot of MAbs reacted with purified orf viral proteins. Purified orf viral proteins were separated on 10% polyacrylamide gels and transferred to PVDF membranes. Blots were probed with 10 hybridoma secreted MAbs (4A6G12, 7E5H2, 6B8A4, 3D9C4, 2F4G6, 8H3G11, 1C10D7, 2A5B9, 8E8D10, 5F2D8), washed, and incubated with goat anti-mouse HRP-conjugated IgG antibody. The membranes were washed and developed using an enhanced chemiluminescent substrate.
Phylogenetic analysis
Phylogenetic analysis was performed as described previously.(10) Using the Neighbour-Joining method, a bootstrap consensus tree was used to represent the evolutionary history of ORFV086.(23,24) The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test is shown next to the branches.(25) Evolutionary analyses were carried out using the MEGA5 software (MEGA, v5)(25,26) and expressed based on the number of nucleotide substitutions per site. The numbers used in phylogenetic trees represent the Geninfo Identifier (GI) sequence identification number in GenBank (
Cross-reactivity experiments of 5F2D8
Reactivity analysis of 5F2D8 with other orf viral proteins, ORFV024 and ORFV121, different strains of ORFV, and other poxviruses was performed by ELISA.(12) Briefly, ELISA plates were coated with ORFV086, ORFV024, and ORFV121 protein of NA1/11 strain and with purified viral proteins of different ORFV strains (NA1/11, HN3, FJ-NP, FJ-FQ) and with purified viral proteins of other poxvirus (Tiantan strain of vaccinia virus, F3 strain of fowl poxvirus, and GTPV-FZ strain of goat poxvirus) (100 μL, 1 μg/mL, 4°C, overnight). They were blocked with 1% BSA (2 h, 4°C) and incubated with 100 μL (1 μg/mL) purified 5F2D8 (1 h, 37°C). The wells were washed with PBS and incubated 30 min with HRP-conjugated goat anti-mouse antibodies (Santa Cruz Biotechnology). The plates were then washed 4 times with PBS and incubated 10 min with ABTS substrate (Roche). The OD405 of the solutions were then measured.
Results
Identification of purified viral proteins by MAb 5F2D8
The viral proteins targeted by MAb 5F2D8 were isolated, separated in 12% SDS-PAGE, and stained with GelCode Blue (Fig. 2A). Western blot of the proteins bound with MAb 5F2D8 detected an approximately 20 kDa protein band (Fig. 2B). Six major bands, three bands between 17 and 28 kDa, one 40 kDa, one 70 kDa, and one 100 kDa, corresponding in size to that of Western blot analysis, were excised for microsequencing (Fig. 2A). A total of six major immunoprecipitated proteins in the gel were excised, digested, eluted, and submitted to mass spectrometry analysis for peptide identification.
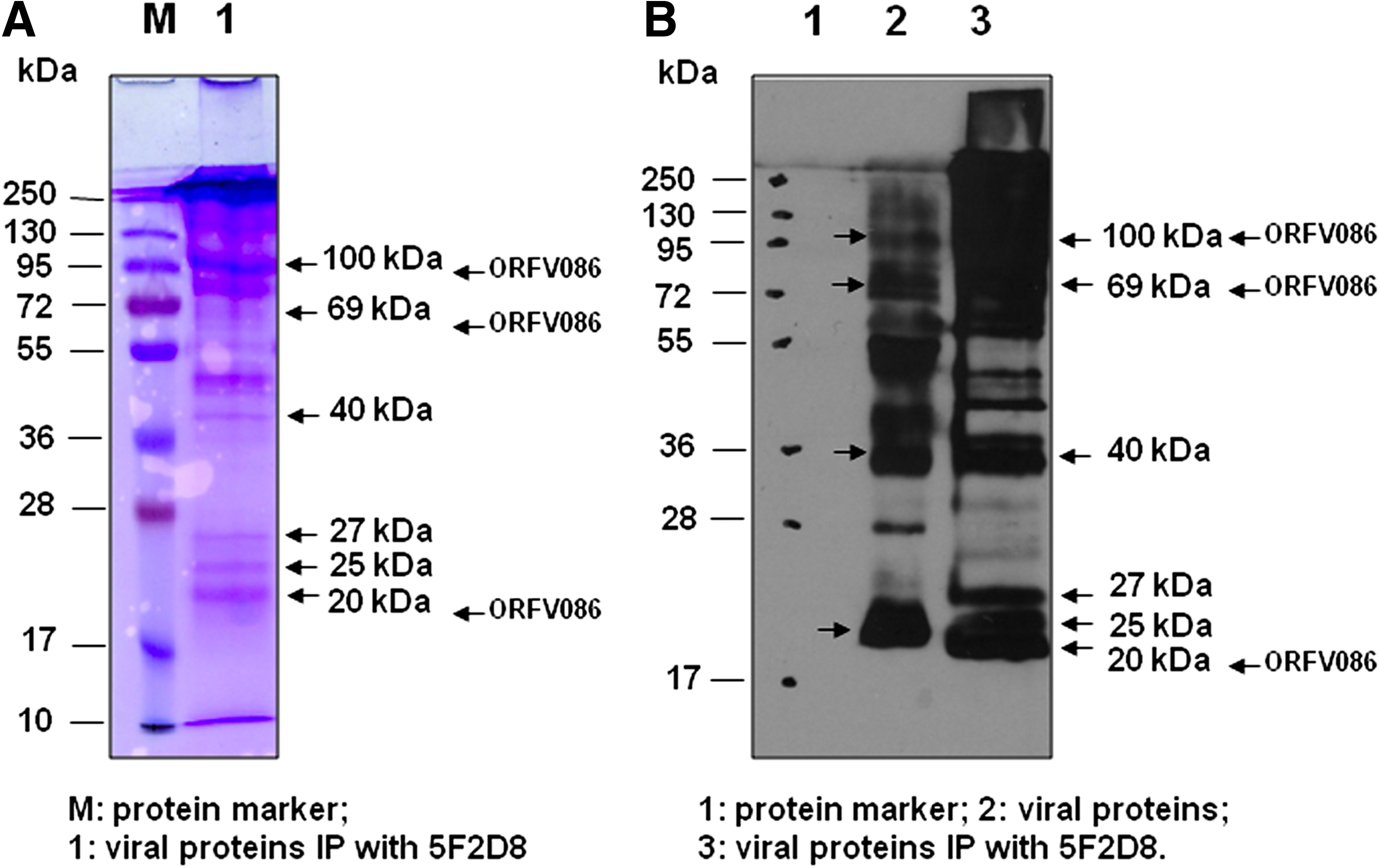
Immunoprecipitation of purified viral proteins with MAb 5F2D8. Monoclonal antibody (MAb) 5F2D8 was incubated with purified viral proteins (O/N, 4°C). Protein G beads were added into the Ag-Ab mixtures to perform immunoprecipitation. (
The peptide sequences were aligned to the predicted amino acid sequences from NCBInr and a total of 11 fragments with protein score 56 (greater than significance score 44 [p<0.05]) were matched to ORFV086 protein (gi|41057149|ref|NP_957863.1|) (Fig. 3, matched peptides shown in bold blue or red). Band 2 with protein score 33.4 was a non-significant protein, and band 3 was matched to the light chain of IgG, respectively. That the molecular weight of the determined protein (20 kDa or 70 kDa) was manifestly smaller than that of the predicted protein (100.05 kDa) encoded by ORFV086 indicates that either the 20 kDa or 70 kDa protein was just one fragment of the ORFV086 precursors, and this is consistent with the 62, 23, and 9 kDa products released by vaccinia virus P4a.
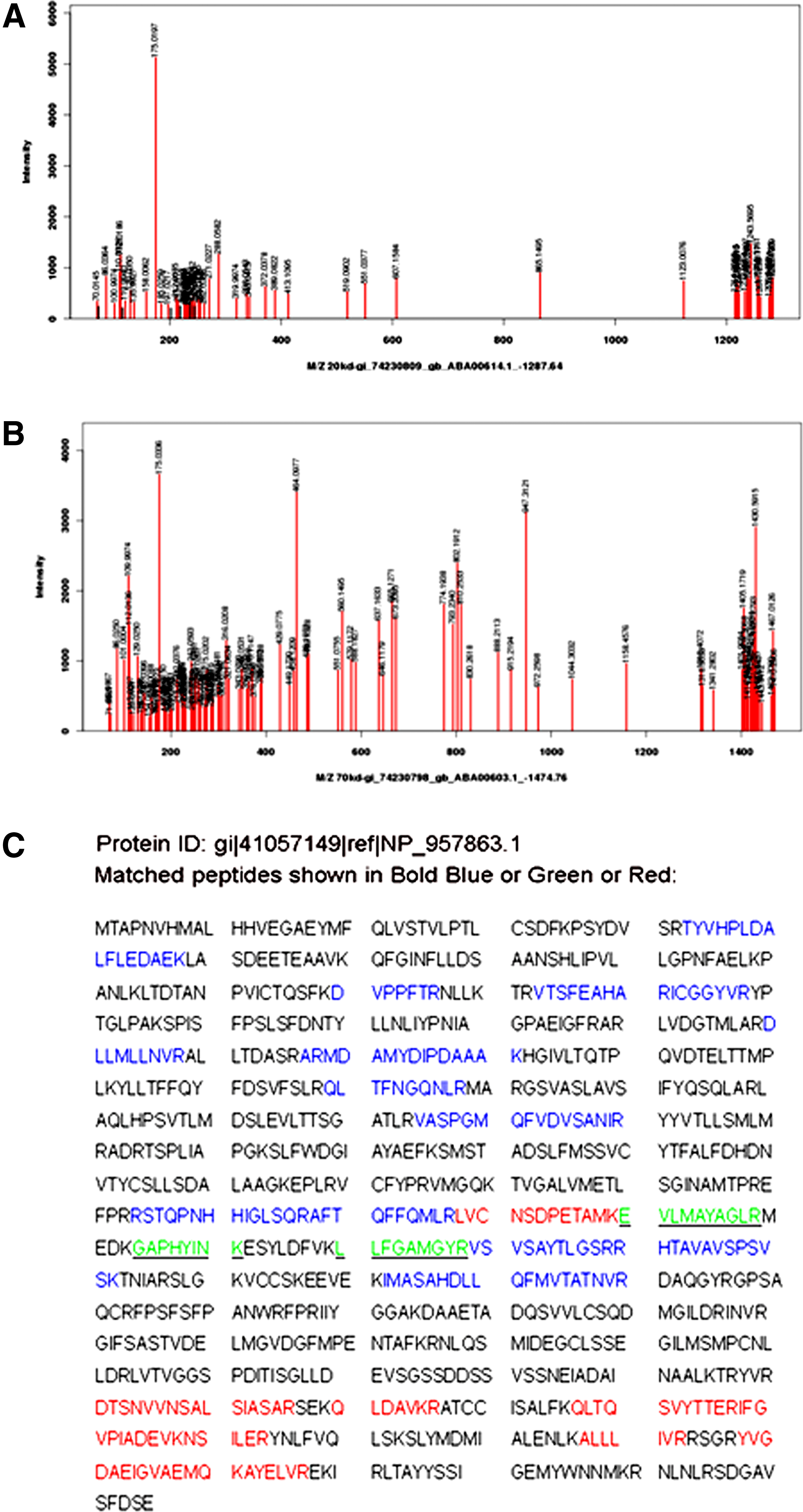
Microsequence analysis of viral proteins immunoprecipitated with MAb 5F2D8. Gelspots were digested with trypsin, followed by peptide elution, and mass spectrometry analysis for identification. (
Immunoflourescent detection of ORFV086 protein with MAb 5F2D8
To determine the utility of MAb 5F2D8 for immunodetection of the ORFV086 protein in infected mammalian cells, OFTu monolayers were infected with NA1/11 and fixed at various times post-infection. The permeabilized cells were incubated with MAb 5F2D8 and then with Alex Fluor 488-conjugated goat-anti-mouse IgG antibodies (Fig. 4A, column ORFV086, green). To visualize cell nuclei and virus factories, the cells were counterstained with DAPI (Fig. 4, DAPI columns, blue images). As shown in Figure 4A (merge columns), MAb 5F2D8 was reactive only in virus-infected cells, as evidenced by the presence of virus factories within the cytoplasmic region and the sites of virus DNA replication and assembly. Additionally, the fluorescent signal was observed only after 12 h post-infection. This suggests that ORFV086, a late-expressed protein, became localized exclusively in virus factories. Based on the time-dependent, immunofluorescent detection of the ORFV086 protein in infected OFTu cells, the gene responsible for this protein is likely transcribed late during the replication cycle of the virus. This delayed expression of the ORFV086 gene was also seen in Western blot analysis of infected cell lysates (Fig. 4C). In these cases, only those samples prepared at 24 h post-infection (not at 4, 8, or 12 h p.i.) contained the ORFV086 protein, while the housekeeping GAPDH protein was readily found in all cases.
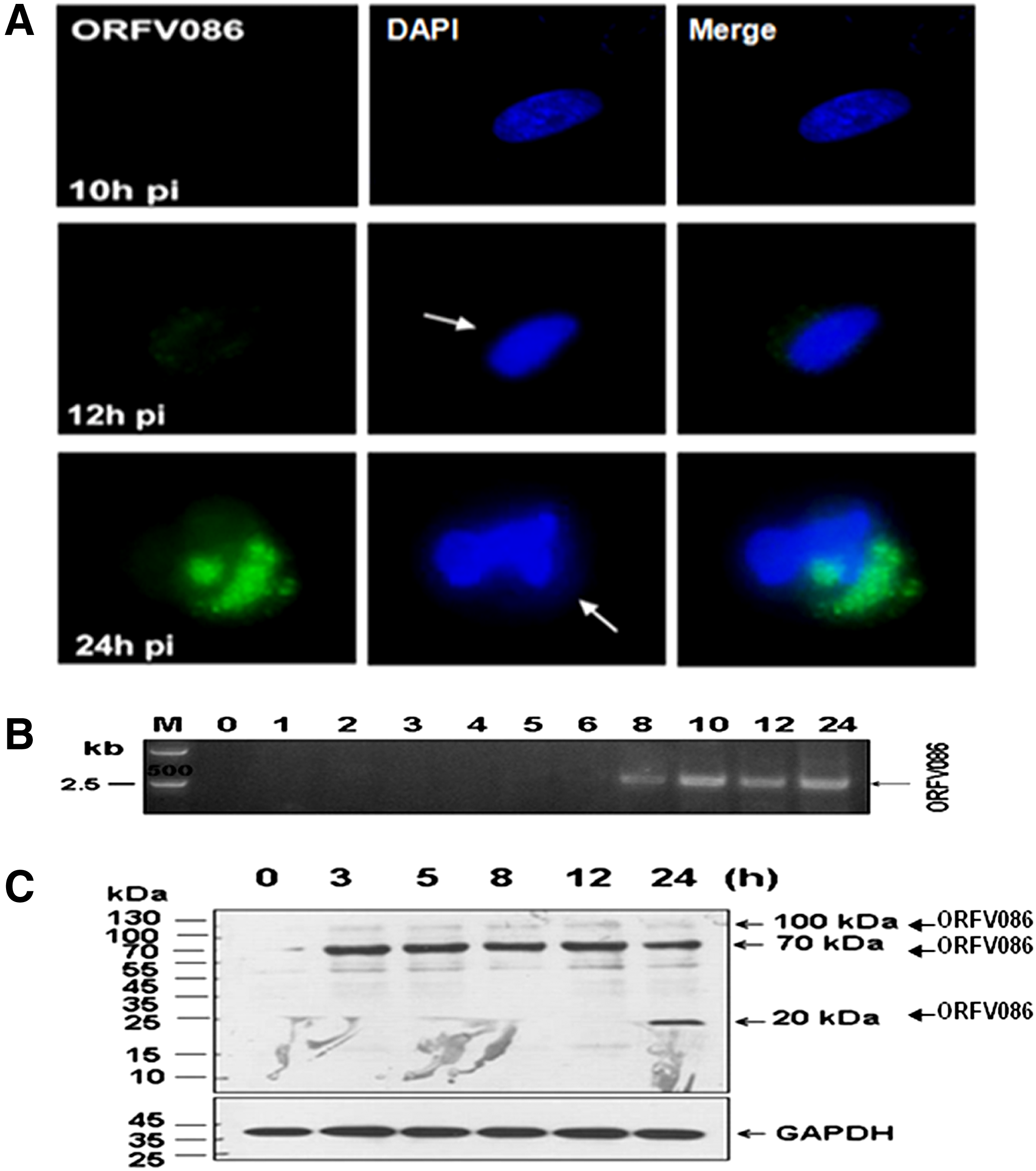
Immunofluorescent detection and temporal expression of intracellular ORFV086 protein. (
Neutralization of ORFV-Jilin infectivity by MAb 5F2D8
The ORFV086 protein is involved in virus adsorption to host cells.(28–30) Because MAb 5F2D8 binds to this protein, it could, theoretically, neutralize ORFV. Using a modified fluorescent focus neutralization assay, the relative infectivity of an NA1/11 virus preparation was measured in vitro after incubation with MAb 5F2D8. With the untreated virus (NMS) as a control, virus neutralization activity of 5F2D8 was shown by the reduction of infectivity of the virus (Fig. 5A). Statistically significant decreases (p<0.001) in virus infectivity were seen after pre-exposure of the virus to the lowest dilution of MAb compared to using normal mouse serum as a negative control (Fig. 5A). The above assay was repeated in triplicate, obtaining the same results, which indicates that 5F2D8 possesses ORFV neutralizing capability.
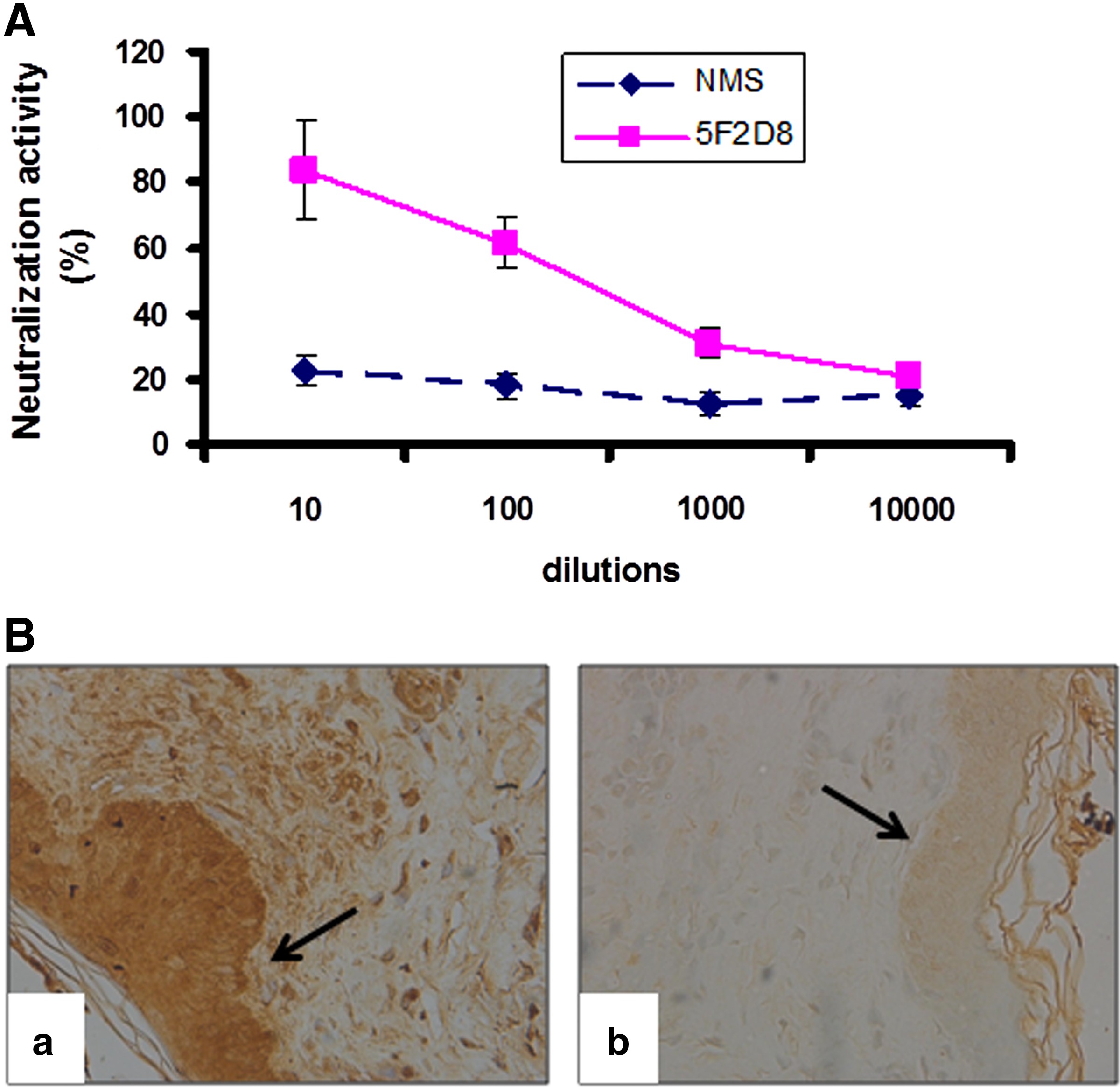
(
Immunohistochemical detection of ORFV-Jilin with MAb 5F2D8
As it was shown to be suitable for detecting ORFV proteins, MAb 5F2D8 was used to examine the distribution of virus in skin lesions from a sheep suffering from orf infection. Sectioned tissue was isolated and processed in immunohistochemical analysis, utilizing MAb 5F2D8. The virus was found in high concentrations in kerotinacytes, developed cells present in the epidermal layer, and invasive inflammatory cells in the subcutaneous tissues (Fig. 5B). However, subcutaneous connective tissues were devoid of virus. It should be noted that the observed virus localizations appeared to be genuine, as replacement of the MAb with normal mouse serum precluded any positive reactions in the tissue sections (Fig. 5B).
Characterization of ORFV NA1/11 ORFV086 protein and phylogenetic analysis
PCR amplification resulted in an amplicon with a 2,715nt open reading frame corresponding to the ORFV-NA1/11 ORF086 gene. NA1/11 had the following nine amino acid substitutions (identity is 99.23%), V87I, T101A, N232H, A237T, I335V, A355T, T373A, G526V, and A759T, compared to NZ2 (Fig. 6). In the phylogenetic tree of ORFV086, NA1/11 clusters together with NZ2 and IA82 (Fig. 7). Nucleic acid and amino acid sequence alignments of the NA1/11 strain ORFV086 gene with the NZ2 strains and IA82 strains showed that the sequences of nucleic acids and amino acids were 99% identical. Comparing the corresponding ORFV086 protein of other strains (OV-IA82, NZ2, OV-SA00, D1701), the results demonstrate that ORFV086 is a conserved gene with 99% identity (Fig. 8). Furthermore, the comparison of the ORFV059 gene (97% identity) (Fig. 9A) and ORFV080 gene (90% identity) (Fig. 9B) of different strains (OV-IA82, NZ2, OV-SA00, D1701) shows more variation than the ORFV086 gene. These results demonstrate the superiority of ORFV086 over the other genes for detection, identification, and phylogenetic analysis.

Comparison of predicted amino acid sequences of ORFV086 protein of NA1/11, NZ2, and Vaccinia virus strain ankara. Predicted amino acid sequences of ORFV086 protein expressed by NA1/11 (AFX81702), NZ2 (DQ184476), and Vaccinia virus Ankara (U94848) strains were based on the nucleotide sequences of corresponding gene, as determined here. Sequences were aligned by using the CLUSTAL W program (San Diego Supercomputer Center biology workbench;

Phylogenetic analysis based on nucleotide sequences of ORFV086. •, NA1/11 isolated in this study. NA1/11 (AFX81702), NZ2 (DQ184476), OV-IA82 (AY386263.1), OV-SA00 (AY386264.1), D1701 (HM133903.1), Pseudocowpox virus strain F00.120R (GQ329669.1), Bovine papular stomatitis virus strain BV-AR02 (AY386265.1), Vaccinia virus Ankara (U94848), Deerpox virus W-848-83 (AY689436.1), Cowpox virus strain UK2000_K2984 (HQ420900.1).

Comparison of the corresponding ORFV086 protein of other strains (OV-IA82, NZ2, OV-SA00, D1701). Predicted amino acid sequences of ORFV086 protein were based on nucleotide sequences of corresponding gene as determined here. Sequences were aligned by using the CLUSTAL W program (San Diego Supercomputer Center biology workbench).

Comparison of predicted amino acid sequences of ORFV059 gene (
Cross-reactivity of 5F2D8
Reactivity of 5F2D8 with different proteins of ORFV and different orf parapoxvirus strains and other poxviruses (Table 1) was observed using ELISA. As shown in Table 1, 5F2D8 can recognize ORFV086 of NA1/11. No cross-reactivity was detected with other viral proteins such as ORFV024, ORFV121. In addition, 5F2D8 also recognizes ORFV086 in various ORFV strains isolated from different regions of China. Tested against other poxviruses, no cross-reactivity was detected (Table 1).
Discussion
Orf is an acutely contagious disease, caused by ORFV, which infects goats, sheep, and humans.(31) The most recent report revealed an increase in infected animals species, including cats, reindeer, camels, and serows, and suggested the expansion of the hosts that could be infected.(32) Owing to the worldwide distribution of ORFV, orf is one of the important epidemic diseases in sheep and exists in almost every sheep and goat raising country.(32) ORFV has a strong resistance to the environment and, once infecting a herd, is not easy to remove. It is an enduring hazard for infected sheep and brings economic loss for animal husbandry. At present, this disease is widespread in some major sheep and/or goat raising countries of Europe, America, and Africa, seriously threatening the development of the sheep/goat industry and human health over the world.(1,33–37) In China, outbreaks of orf in provinces such as Qinghai, Xinjiang, Inner Mongolia, Jilin, and Heilongjiang(10,38) severely impact the sheep industry. This problem is most severe in Jilin Province.(10) Therefore, this disease attracts great attention there.
The ORFV086 gene has been found to encode a structural protein expressed on the intracellular virion core; it is similar to the VV P4a and other poxvirus homologues.(13,14) The VV P4a is the most abundant protein, accounting for about 14% of the virion.(39) The proteolysis of P4a has an important role in progeny virus particle assembly, morphogenesis, and maturity.(14,15,40) It is generally believed that the vaccinia virus assembly and morphogenesis process are as follows: Shortly after DNA replication begins, electron dense areas form in the cytoplasm, which are designated as virus factories or virosomes.(40–42) Assembly of the virus begins at virosomes surrounded by crescent membranes, which later engulf granular materials and form immature virions (IV). The IVs transform into infectious structures referred to as intracellular mature virions (IMV). Proteolytic processing of vaccinia virus core proteins appears to be essential in the formation of IMV. A portion of the IMV then is developed into intracellular enveloped virus (IEV). Following migration to the cell surface, the outermost IEV membrane fuses with the plasma membrane to give rise to extracellular enveloped virus (EV). The EV can either remain associated with the cell (cell-associated enveloped virus, CEV) or be released into the external medium as extracellular enveloped virus (EEV).(43)
The proteolysis of VV core proteins occurs only in the process of transforming IV to IMV, making the spherical-shaped IV into a brick-shaped IMV. P4a, a 102 kDa protein encoded by A10L, is synthesized late in the viral infection. This product is post-translationally cleaved into three smaller polypeptides (62, 23, and 9 kDa products).(13,14,44,45) The 23 kDa protein and the 4a (62 kDa) protein become major components of the virus, while the fate of the 9 kDa peptide is unknown. In this study, immunofluorescent assays found that the ORFV086 protein was located in the “virus factory” and could be observed as early as 12 hours p.i., which indicated ORFV086 is a late gene product. Immunoprecipitation and microsequencing assays demonstrated that the 19 kDa and 70 kDa are derived from ORFV086. Our observation that the sequences of 19 kDa and 70 kDa overlapped between Glu-490 and Arg-590 suggests that the potential cleavage sites may localize in this area (Fig. 3).
Mechanisms of host immunity to orf virus infection are not well understood and protective antigens need to be identified. Normally, orf is diagnosed based on pathology and clinical symptoms.(46) However, it may be difficult to recognize the early stages of infection. Therefore, antibodies, such as monoclonal antibodies, which are highly specific, are of particular value in orf research and diagnosis.(47)
As shown in the results section, monoclonal antibody 5F2D8 is a useful tool for diagnostic and epidemiological studies using Western blot, ELISA, and immunohistochemical assays, exploring the mechanism of ORFV pathogenesis by determining virus entry and location using immunohistochemical, immunoprecipitation, and immunofluorescent techniques.(1–3,5,9,46) This monoclonal antibody may also be utilized for development of a subunit vaccine and in therapeutic medicine.(47)
We demonstrated that MAb 5F2D8 produced in this study is specific against ORFV086 protein. Transcription profile and Western blot analysis showed that the ORFV086 gene encodes a structural protein that is expressed on the intracellular, mature virion core and has structural similarity with the VV precursor core protein P4a and other poxvirus homologues.(13–15) ORFV086 gene of NA1/11 strains is 2718 bp long, encoding a 100.05 kDa protein. ORFV086 is centrally located in the genome, is highly conserved, and plays a key role in viral replication and morphogenesis. Nucleic acid and amino acid sequence alignment of ORFV086 between NA1/11 and NZ2 strain showed that both of these were 99% identical. NA1/11 ORFV086 had the following nine amino acid substitutions: V87I, T101A, N232H, A237T, I335V, A355T, T373A, G526V, and A759T (Fig. 6). Phylogenetic analysis indicates the ORFV086 of NA1/11, NZ2, and OV IA82 strains has high homology (Fig. 7). Amino acid sequence alignment of NA1/11and NZ2 ORFV086 with vaccinia virus core protein precursor P4a found that the predicted proteolytic sites of ORFV086 proteins are similar, yet distinct from precursor P4a. The P4a precursor is cleaved at two locations, the Ala-Gly-Ser site (residues 613 to 615) and the Ala-Gly-Thr site (residues 696 to 698). Unlike P4a, ORFV086 was predicted to cleave at two distinct locations, the GGA site (residues 621 to 623) and the GGS site (residues 708 to 710). However, exact proteolytic sites of NA1/11 ORFV086 need further verification.
In this study, the purified orf viral proteins were used to inoculate mice and produce monoclonal antibodies. We identified anti-ORFV protein MAb 5F2D8 via an indirect ELISA, and selected it for its high specificity. After immunoprecipitation and microsequencing, 5F2D8 was found to specifically recognize NA1/11 ORFV086. Notably, 5F2D8 has neutralization activity. Immunofluorescent and Western blot analyses demonstrated that ORFV086 is a late-expression gene. Therefore, 5F2D8 may be useful for future research into the mechanism of ORFV infection and the development of an immunodiagnostic assay and, potentially, a vaccine to prevent infection and control orf disease.
Footnotes
Acknowledgments
This study was supported by grants (nos. 31070138 and 31170147) from the National Natural Science Foundation of China (NSFC), the Scientific Research Foundation of Introducing Talents of Southern Medical University (2010), and Special Funds for Colleges and Universities Talents of Guangdong Province (2011).
Author Disclosure Statement
The authors have no financial interests to disclose.