
Abstract
Monoclonal antibodies are known to have several applications in clinical diagnosis and therapy. In the present study, the truncated S1 gene, encoding the exterior of the viral spike protein of porcine epidemic diarrhea virus (PEDV), was subcloned into prokaryotic expression vector pET32a (+) and expressed as a recombinant protein in Escherichia coli BL21(DE3). Female BALB/c mice were immunized with the purified recombinant truncated S1 protein, and three monoclonal antibodies (MAb designated as E3, G8, and G9) against the truncated S1 protein obtained by hydridoma technique. Further characterization demonstrated that the three MAbs (E2, G8, and G9) belong to IgG1 subclass and have different affinities (G9 > G8 > E3). Furthermore, all of the three MAbs reacted with PEDV in the fluorescent antibody assay. Our study suggests that purified truncated S1 protein and the three developed MAbs could be useful in the development of a diagnostic assay for anti-PEDV antibodies and PEDV antigen, respectively.
Introduction
P
Monoclonal antibodies (MAbs) are widely used in clinical diagnosis and therapy. MAbs have been applied to detect virus or antibody in different assays, including fluorescent antibody test and ELISA.(15–17) Previous studies showed that there were multiple antigenic epitopes in the amino-terminal of S1 gene, and polyclonal antibodies against this region can significantly inhibit the proliferation of the virus.(14) In the present study, a truncated S1 recombinant protein was expressed in Escherichia coli and used to immunize female BALB/c mice. Three MAbs were achieved by hybridoma technique and further characterized.
Materials and Methods
Cloning of truncated S1 gene (tS1 gene)
Viral RNA was extracted from PEDV SDBZ strain isolated from a clinical sample by the RNA genome extraction kit. One pair of spike gene primers, sense primer 5'-GGC
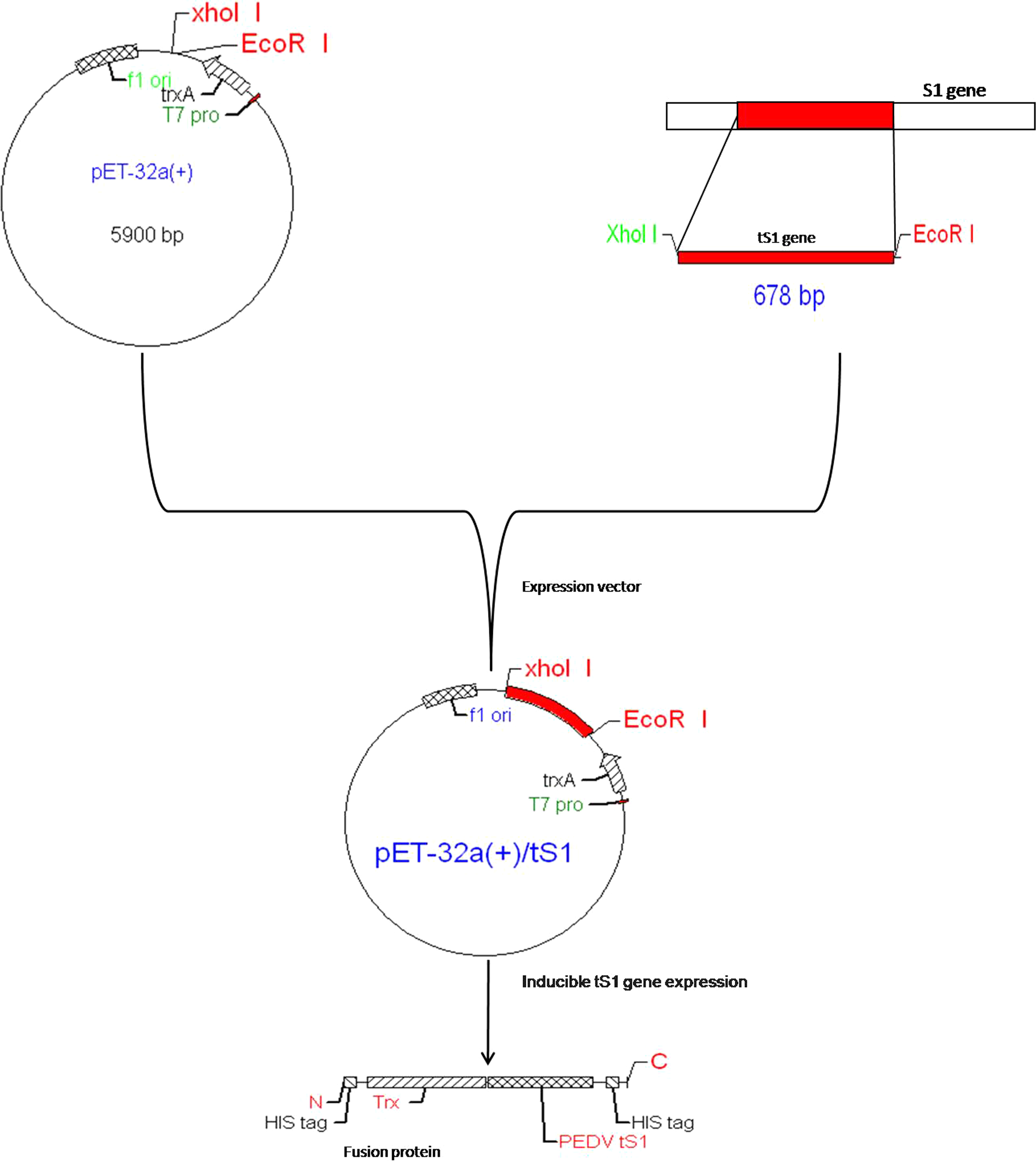
Schematic showing strategy used for cloning of PEDV tS1 gene into expression vector pET32a(+).
Preparation of recombinant protein tS1
E. coli BL21(DE3) were transformed with pET32a(+)/tS1 plasmid and cultured in LB medium (ratio of 1:100). Cells were induced with 1 mM of isopropyl-β-D-thiogalactopyranoside (IPTG) for 4 h at 37°C, 225 rpm. The cells were collected by centrifugation and re-suspended in appropriate amount of PBS (pH 7.4) buffer and sonicated. After high-speed centrifugation, tS1 protein in the supernatant and sediment were detected by the SDS-PAGE electrophoresis. The protein was further purified with AKTApurifier100 (GE Healthcare, Little Chalfont, United Kingdom) after dialysis and filtration. The concentration of purified protein was detected by chromatograph Bradford assay and SDS-PAGE protein electrophoresis.
Preparation of MAb anti-tS1 protein
Immunization of mice
Four, 6-week-old, female BALB/c mice were immunized with tS1 protein (60 μg/mouse) mixed with equal amount of Freund's complete adjuvant and boosted twice with the tS1 protein (30 μg/mouse) mixed with equal amounts of Freund's incomplete adjuvant at bi-weekly intervals. One week after the third injection, mouse antisera were collected and tested for the titer of antibody. Mice induced with higher antibody titers were boosted 3 days before hybridoma production.
Cell fusion and screening
A cell fusion procedure was done as previously described.(18) The antibody was screened by indirect ELISA. Briefly, polystyrene microtiter plates were coated with 100 μL of tS1 protein (2 μg/mL) in 0.05 M carbonate buffer (pH 9.6) overnight at 4°C and were washed twice with 200 μL of phosphate-buffered saline with 0.05% Tween-20. The plates were blocked with 200 μL of 2% dry milk in 0.01 M PBS at 37°C for 2 h. The wells were then washed three times. The washing step was followed after incubation with primary and secondary antibodies as well. 100 μL of fusion cell supernatant from the culture dish wells were dispensed into microtiter plates coated with tS1 protein at 37°C for 1 h. Goat anti-mouse IgG peroxidase conjugate (100 μL at 1:20,000 dilution) was added to each well, and plates were incubated for 1 h at 37°C. To develop color, 100 μL of Ultra TMB was added to each well, and the plates were incubated for 15 min at room temperature followed by the addition of 50 μL of stopping solution (1 N HCl). The OD was determined at 450nm using a plate reader. An OD450 ≥0.3 was considered positive.
Characterization of monoclonal antibodies
The hybrdoma cells were propagated 15 times continuously. Their stability was examined by detecting the antibody titers of cell supernatants using an indirect ELISA method as previously described. Monoclonal immunoglobulin subclass was determined by the mouse MAb isotyping kit. The relative affinity of monoclonal antibodies was determined by using a indirect ELISA method, as previously reported.(19) Briefly, microtiter plates were coated with tS1 protein (2 μg/well) at 4°C overnight. The plates were then washed three times with 200 μL of phosphate-buffered saline with 0.05% Tween-20 (PBS-T) and blocked with 200 μL of 2% dry milk in 0.01 M PBS. After being washed three times with PBS-T, the serial diluted purified MAbs (original concentration of MAbs is 1 mg/mL) were added into the wells and the normal mouse IgG were added as negative controls. All tests were performed in double sets. After incubation for 1 h at 37°C and being washed three times, goat anti-mouse IgG peroxidase conjugate (100 μL at 1:20,000 dilution) was added to each well, and plates were incubated for 1 h at 37°C. After being washed three times, 100 μL of Ultra TMB (Thermo Scientific, Rockford, IL) was added to each well, and the plates were incubated for 15 min at room temperature. The reaction was ended by 100 μL of 1 M HCL, and OD value at 450 nm was detected.
Determination of MAbs reaction with virus
Indirect immunofluorescence test
The Vero cells were first seeded onto 24-well culture plate and then infected with PEDV after overnight, and then cultured in the DMEM at 37°C for 48 h. After removing the supernatant, the cells were fixed in paraformaldehyde for 15 min, treated with 1% Triton x-100 for 15 min, and then washed three times with PBS. Three monoclonal antibodies (E3, G8, G9) were added onto the plate at 37°C for 1 h. After washing three times with PBST (PBS with 0.05% Tween-20), a fluorescent-labeled rabbit anti-mouse secondary antibody (100 μL at 1:100 dilution; Beijing, China) was added into the wells and incubated for 1 h at 37°C, then washed in triplicate; fluorescence signals were observed by fluorescence microscope (CKX41, Olympus, Tokyo, Japan).
Direct immunofluorescent antibody test
The best affinity monoclonal antibody is labeled by fluorescein isothiocyanate (FITC, Sigma, St. Louis, MO). Briefly, monoclonal antibodies (1 mg/mL) were dialyzed against cross-linking reaction buffer (0.1 M NaHCO3, 0.01 M Na2CO3, 0.13 M NaCl [pH 9.0]), conjugated to FITC at a ratio 6.67:1 of monoclonal antibody to FITC at 4°C for 8 h under dark conditions, and purified by gel filtration (Sephadex G75, Sigma) using borate-buffered saline as eluting buffer.(20) The Vero cell were infected with PEDV fixed in paraformaldehyde for 15 min, treated with 1%Triton x-100 for 15 min, then washed three times. The labeled monoclonal antibody was added to the plate and incubated for 30 min in a moist chamber at 37°C. It was then observed with a fluorescence microscope at a magnification of 200X.
Results
Construction of recombinant expression vector
Expression plasmid pET32a (+)-tS1 was successfully constructed and a target band size of 678 base pairs (bp) was detected after digestion by double enzymes (EcoRI and XhoI) (Fig. 2A). Sequencing analysis showed that the sequence of SDBZ isolate is close to the Europe prephnotye strain SM98 (GU937797.1). Moreover, in comparison with DR13 (JQ023161.1), LZC (EF185992.1), and CV777 (AF353511.1) strains, 12 bp insertions and several point mutations were found (Fig. 2B).
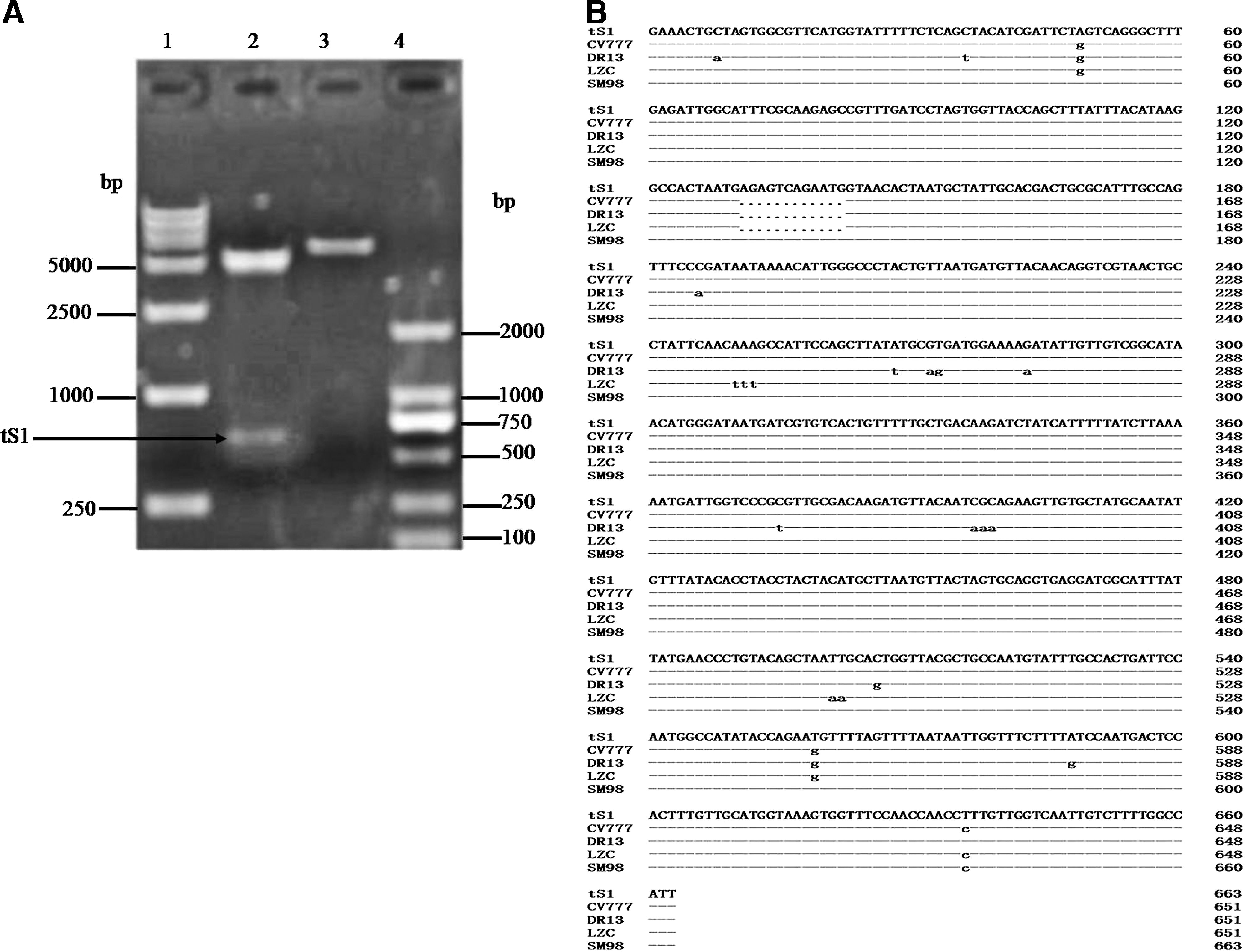
Identification of recombinant expression vector pET32a(+)/tS1. (
Expression and purification of tS1 truncated protein
BL21(DE3) transformed with pET32a(+)-tS1 were induced by 1 mM IPTG for 4 h at 37°C. SDS-PAGE analysis showed that 32 kDa of protein was produced and most of the target protein existed in the inclusion bodies. Western blot assays demonstrated that the expressed tS1 specifically reacted with PEDV positive sera of rabbit (Fig. 3). The protein was purified using Nickel ion affinity chromatography and its concentration was 2.14 mg/mL determined by Bradford method.
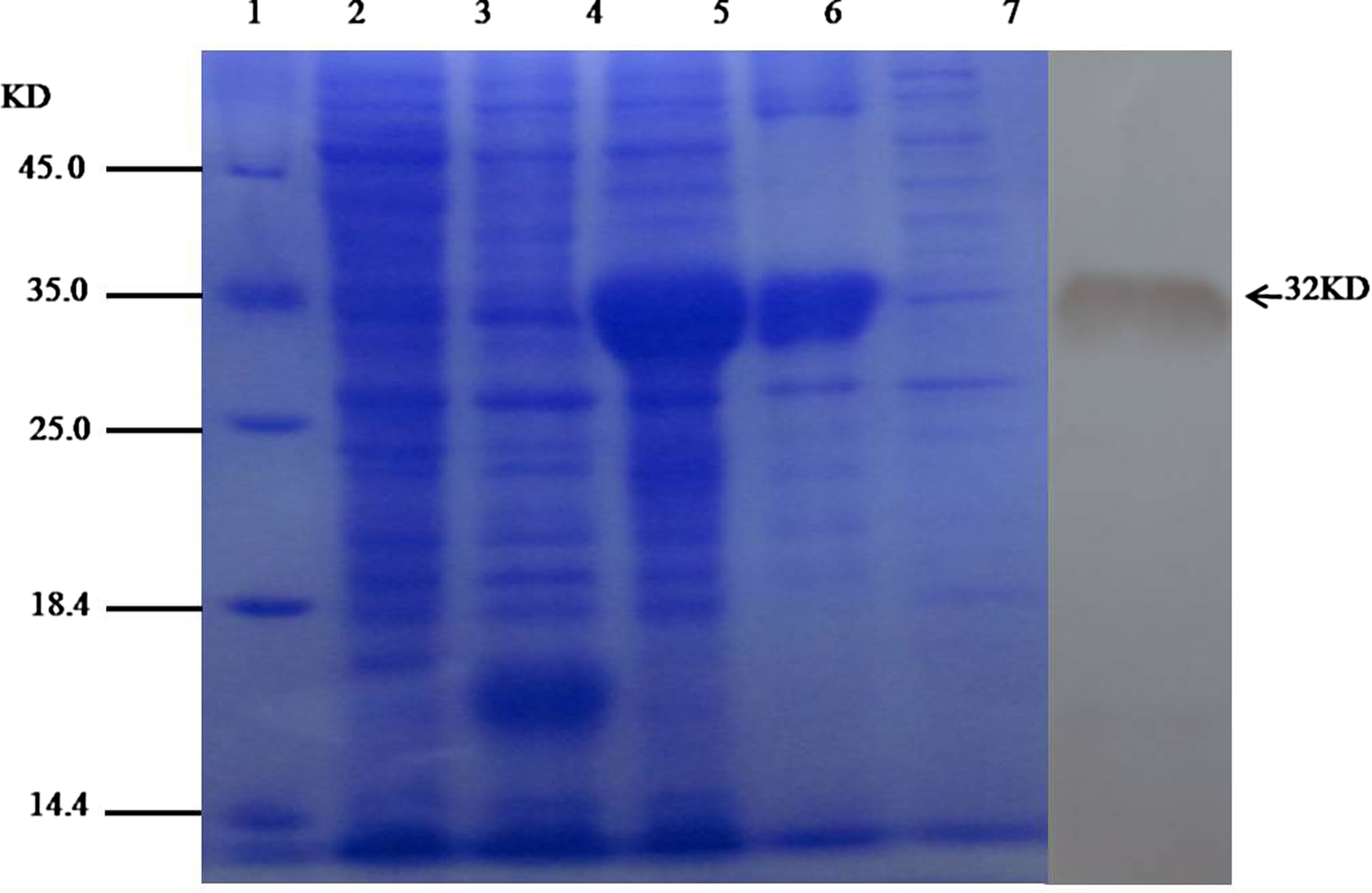
Analysis of recombinant proteins: SDS-PAGE and immunoblot. Lane 1, protein molecular weight markers; lane 2, expression strain without induction; lane 3, empty vector bacteria with induction; lane 4, expression strain with induction, lane 5, precipitation of expression strain bounce broken; lane 6, supernatant of expression strain bounce broken; lane 7, Western blot of expression products.
Production, reactivity, and characterization of monoclonal antibodies
Three monoclonal antibodies (G8, G9, E3) were screened. The titer of three monoclonal antibodies of cell culture supernatants and ascites were 1:1000, 1:2000, 1:2000, and 1:128,000, 1:256,000, 1:256,000, respectively, by the method of indirect ELISA (data not shown). Its culture supernatant titers remained stable when the cells were continuously propagated 15 times. Three monoclonal antibodies also belonged to IgG1 subclass by identification of the kit. Three monoclonal antibodies were purified by the method of protein G column. The concentrations were 2.51 mg/mL, 2.56 mg/mL, and 4.41 mg/mL, respectively. The standard curves were drawn by competitive ELISA, which showed that the size of relative affinity were G9>G8>E3 (Fig. 4). Indirect immunofluorescence tests showed there was a good reactivity between the three monoclonal antibodies and PEDV (Fig. 5A). This study labeled G9 monoclonal antibody by FITC as the direct fluorescent antibody; the result showed that it can detect the PEDV after 100-fold dilution (Fig. 5B).

Monoclonal antibody affinity constant of detection with ELISA method.

Results of IFA detection of PEDV (200x). (
Discussion
Monoclonal antibodies have many applications in biology and medicine, including enzyme-linked immunosorbent assay (ELISA), immunohistochemistry assay (IHA), radioimmunoassay analysis, and flow cytometry techniques.(15–17,21,22) Selection of immunogen is key to the successful development of highly specific monoclonal antibodies; therefore, this study aims to select highly immunogenic antigen by utilizing different approaches.(23)
First, this study predicted the major antigenic region using software (DNA Star, Madison, WI). The region affected PEDV multiplication was determined, which provided a theoretical basis to achieve the desired goal and laid the foundation for high-quality selection of immunogen; secondly, the study used pET32a(+) vector containing His-tag to express truncated S protein. The molecular weight of the His-tag is smaller than that of GST-tag, reducing the influence of the spatial structure of the expression protein and also facilitating the purification of the target protein, which ensured a high purity protein.
Finally, anti-truncated S protein rabbit serum was produced. The anti-truncated S protein rabbit serum could inhibit the PEDV proliferation in Vero cells, indicating that the expressed protein was an important portion of the S protein responsible for inducing neutralizing antibodies.
Currently, the rapid detection methods of PEDV mainly include PCR, ELISA, and colloidal gold strip.(24–26) The advantages of these methods are speed and accuracy, but there is a higher requirement for the laboratory environment and operating personnel. The present study showed that the screening monoclonal antibodies have good reactogenicity and can react with not only the immunogen but also the whole virus, which can be useful for development of direct immunofluorescence technique and colloidal detection technology of PEDV to enrich the existing rapid detection PEDV method. At the same time, future studies will determine if the three monoclonal antibodies can inhibit the proliferation of PEDV in the Vero cells or under other conditions. Our study indicates that the monoclonal antibodies developed are useful in researching virus epitopes, infected mechanisms, and new therapeutic agents.
Footnotes
Acknowledgments
This research is supported by the Postdoctoral Fund of Shandong Binzhou Animal Science and Veterinary Medicine Academy Postdoctoral Research Station (201201) and the Postdoctoral Innovation Fund of Shandong Province (201303027).
Author Disclosure Statement
The authors have no financial interests to disclose.