
Abstract
Podoplanin (PDPN) is a type-I transmembrane sialoglycoprotein that possesses a platelet aggregation-stimulating (PLAG) domain in the N-terminus. PLAG domain includes three tandem repeats of eight amino acids: PLAG1, PLAG2, and PLAG3. Among the three PLAG domains, O-glycan on Thr52 of PLAG3 is critical for binding with C-type lectin-like receptor-2 (CLEC-2) and is essential for platelet-aggregating activity of PDPN. In contrast, the glycosylation of Thr34 of PLAG1 of human PDPN remains to be clarified. Herein, we developed and characterized a novel anti-PDPN monoclonal antibody, LpMab-10, which targets PLAG1/2 domain. LpMab-10 detects endogenous PDPN of cancer cells and normal cells independently of glycosylation. The minimum epitope of LpMab-10 was identified as Glu33–Gly45 of PDPN using Western blot and flow cytometry. The Thr34 of PLAG1 is critical for LpMab-10 recognition, and O-glycan is not included in LpMab-10 epitope, indicating that Thr34 of PLAG1 is not O-glycosylated. In immunocytochemical and immunohistochemical analyses, LpMab-10 strongly detected PDPN-expressing tumor cells. By using monoclonal antibodies against different Ser/Thr, including epitopes of PDPN, it becomes possible to determine whether Ser/Thr residues of PDPN are O-glycosylated.
Introduction
P
Materials and Methods
Cell lines, animals, and tissues
LN229, NCI-H226, HEK-293T, Chinese hamster ovary (CHO)-K1, glycan-deficient CHO cell lines (Lec1, Lec2, and Lec8), Met-5A, and P3U1 were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Human lymphatic endothelial cells (LEC) were purchased from Cambrex (Walkersville, MD). The human glioblastoma cell line LN319 was donated by Dr. Webster K. Cavenee (Ludwig Institute for Cancer Research, San Diego, CA). PC-10 cells were purchased from Immuno-Biological Laboratories (Gunma, Japan). LN229, CHO-K1, Lec1, Lec2, and Lec8 were transfected with human PDPN plasmids (LN229/hPDPN, CHO/hPDPN, Lec1/hPDPN, Lec2/hPDPN, Lec8/hPDPN) using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer's instructions.(25) LN319/PDPN-knock out (KO) cells were produced by transfecting CRISPR/Cas plasmids, which targets PDPN (Sigma-Aldrich, St. Louis, MO), using a Gene Pulser Xcell electroporation system (Bio-Rad Laboratories, Philadelphia, PA). CHO-K1, Lec1, Lec2, Lec8, NCI-H226, PC-10, and P3U1 were cultured in RPMI 1640 medium (Nacalai Tesque, Kyoto, Japan), and LN229, LN319, and Met-5A were cultured in Dulbecco's Modified Eagle's Medium (DMEM) medium (Nacalai Tesque), supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific), 2 mM L-glutamine (Thermo Fisher Scientific), 100 U/mL of penicillin, and 100 μg/mL of streptomycin (Thermo Fisher Scientific) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. L-proline (0.04 mg/mL) was added for Lec1, Lec2, and Lec8. LEC was cultured in endothelial cell medium EGM-2MV supplemented with 5% FBS (Cambrex). MITO + serum extender (Thermo Fisher Scientific) was added for Met-5A cells. Female BALB/c mice (4 weeks old) were purchased from CLEA Japan (Tokyo, Japan). Animals were housed under pathogen-free conditions. The Animal Care and Use Committee of Tohoku University approved the animal experiments described herein. This study examined one seminoma patient(11) and one osteosarcoma patient(26) who underwent surgery at Yamagata University Hospital. The ethical committee of the Yamagata University Faculty of Medicine approved our study. Informed consent for obtaining samples and for subsequent data analyses was obtained from patients or the patient's guardian.
Hybridoma production
BALB/c mice were immunized by intraperitoneal (i.p.) injection of 1 × 108 LN229/hPDPN cells together with Imject Alum (Thermo Fisher Scientific). After several additional immunizations, a booster injection was given i.p. 2 days before spleen cells were harvested. The spleen cells were fused with P3U1 cells using PEG1500 (Roche Diagnostics, Indianapolis, IN). The hybridomas were grown in RPMI medium with hypoxanthine, aminopterin, and thymidine selection medium supplement (Thermo Fisher Scientific). The culture supernatants were screened using enzyme-linked immunosorbent assay (ELISA) for binding to recombinant human PDPN purified from LN229/hPDPN cells.
Enzyme-linked immunosorbent assay
Purified proteins or peptides were immobilized on Nunc Maxisorp 96-well immunoplates (Thermo Fisher Scientific) at 1 μg/mL for 30 min. After blocking with SuperBlock T20 (PBS) Blocking Buffer (Thermo Fisher Scientific), the plates were incubated with culture supernatant or purified MAbs (1 μg/mL) followed by 1:1000 diluted peroxidase-conjugated anti-mouse IgG or anti-rat IgG (Dako, Glostrup, Denmark). The enzymatic reaction was conducted with a 1-Step Ultra TMB-ELISA (Thermo Fisher Scientific). The optical density was measured at 655 nm using an iMark microplate reader (Bio-Rad Laboratories). These reactions were performed with a volume of 50 μL at 37°C.
Western blot analyses
Cell lysates (10 μg) were boiled in SDS sample buffer (Nacalai Tesque). The proteins were electrophoresed on 5–20% polyacrylamide gels (Wako Pure Chemical Industries, Osaka, Japan) and were transferred onto a PVDF membrane (EMD Millipore, Billerica, MA). After blocking with SuperBlock T20 (PBS) Blocking Buffer, the membrane was incubated with primary antibodies (1 μg/mL) and then with peroxidase-conjugated secondary antibodies (Dako; 1:1000 diluted), and developed with the Pierce Western Blotting Substrate Plus (Thermo Fisher Scientific) using a Sayaca-Imager (DRC, Tokyo, Japan).
Flow cytometry
Cell lines were harvested by brief exposure to 0.25% Trypsin/1 mM EDTA (Nacalai Tesque).(29) After washing with phosphate buffered saline (PBS, Nacalai Tesque), the cells were treated with primary antibodies (1 μg/mL) for 30 min at 4°C, followed by treatment with Oregon Green 488 goat anti-mouse IgG (Thermo Fisher Scientific). Fluorescence data were collected using a Cell Analyzer EC800 (Sony Corp., Tokyo, Japan).
Determination of binding affinity using flow cytometry
LN319 and PC-10 (2 × 105 cells) were resuspended with 100 μL of serially diluted antibody (0.02–100 μg/mL) followed by secondary anti-mouse IgG (Thermo Fisher Scientific). Fluorescence data were collected using the EC800 cell analyzer. The dissociation constants (KD) were obtained by fitting the binding isotherms using the built-in one-site binding models in Prism software.
Lectin microarray
PDPN from CHO/hPDPN cells was solubilized using 1% Triton-X100 in PBS (PBST) and was purified using a FLAG-tag system (Sigma-Aldrich). Then, 100 μL of purified PDPN (31.25–2000 ng/mL) was applied to a lectin array (LecChip, v1.0; GlycoTechnica, Hokkaido, Japan), including triplicate spots of 45 lectins in each of seven divided incubation baths on the glass slide. After incubation at 20°C for 17 h, 4 μL of human IgG (5 mg/mL; Sigma-Aldrich) was applied to each well. After incubation at 20°C for 30 min, the glass slide was washed three times with PBST; 60 μL of biotinylated LpMab-10 (1 μg/mL) in PBS was applied to the array and then incubated at 20°C for 3 h. After washing three times with PBST, Cy3-labeled streptavidin (Thermo Fisher Scientific) was added to the array and incubated at 20°C for 30 min. The glass slide was scanned using a GlycoStation Reader 1200 (GlycoTechnica). Abbreviation of lectins: LTL, Lotus tetragolonobus lectin; PSA, Pisum sativum agglutinin; LCA, Lens culinaris agglutinin; UEA I, Ulex europaeus agglutinin I; AOL, Aspergillus oryzae L-fucose-specific lectin; AAL, Aleuria aurantia lectin; MAL I, Maackia amrensis lectin I; SNA, Sambucus nigra lectin; SSA, Sambucus sieboldiana agglutinin; TJA-I, Trichosanthes japonica agglutinin I; PHA-L, Phaseolus vulgaris leucoagglutinin; ECA, Erythrina cristagalli agglutinin; RCA120, Ricinus communis agglutinin 120; PHA-E, Phaseolus vulgaris erythroagglutinin; DSA, Datura stramonium agglutinin; GSL-II, Griffonia simplicifolia lectin II; NPA, Narcissus pseudonarcissus agglutinin; ConA, Concanavalin A; GNA, Galanthus nivalis agglutinin; HHL, Hippeastrum hybrid lectin; ACG, Agrocybe cylindricea galectin; TxLCI, Tulipa gesneriana lectin; BPL, Bauhinia purpurea alba lectin; TJA-II, Trichosanthes japonica agglutinin; EEL, Euonymus europaeus lectin; ABA, Agaricus bisporus agglutinin; LEL, Lycopersicon esculentum lectin; STL, Solanum tuberosum lectin; UDA, Urtica dioica agglutinin; PWM, pokeweed mitogen; PNA, peanut agglutinin; WFA, Wisteria floribunda agglutinin; ACA, Amaranthus caudatus agglutinin; MPA, Maclura pomifera agglutinin; HPA, Helix pomatia agglutinin; VVA, Vicia villosa agglutinin; DBA, Dolichos biflorus agglutinin; SBA, soybean agglutinin; PTL I, Psophocarpus tetragonolobus lectin I; MAH, Maackia amurensis hemagglutinin; WGA, wheat germ agglutinin; GSL-I, Griffonia simplicifolia lectin I.
Production of PDPN mutants
The amplified human PDPN cDNA was subcloned into a pcDNA3 vector (Thermo Fisher Scientific), and a FLAG epitope tag was added at the C-terminus. Substitution of amino acids to alanine or glycine in PDPN was performed using a QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). CHO-K1 cells were transfected with the plasmids using a Gene Pulser Xcell electroporation system (Bio-Rad Laboratories).
Immunocytochemical analyses
Cultured cells were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 15 min at room temperature. Cells were then treated with 10% normal goat serum in PBS (NGS/PBS) to block nonspecific binding sites, and were incubated with 1 μg/mL of LpMab-10 or control (PBS) for 1 h at 4°C in a moist chamber. They were incubated with goat anti-mouse IgG-Alexa 488 (dilution 1:200; Thermo Fisher Scientific) for 30 min at room temperature. Cells were also treated with DAPI (Thermo Fisher Scientific) to stain the cell nuclei. They were examined using confocal laser-scanning microscopy (LSM700; Carl Zeiss, Jena, Germany).
Immunohistochemical analyses
Four-μm-thick histologic sections were deparaffinized in xylene and rehydrated. They were then autoclaved in citrate buffer (pH 6.0; Dako) for 20 min. Sections were incubated with 5 μg/mL of primary antibodies overnight at 4°C followed by treatment with Envision+ kit (Dako). Color was developed using 3,3-diaminobenzidine tetrahydrochloride (DAB, Dako) for 10 min; then the sections were counterstained with hematoxylin (Wako Pure Chemical Industries).
Results
Development and characterization of novel anti-PDPN MAb LpMab-10
We immunized mice with LN229/hPDPN cells to develop novel anti-PDPN MAbs. Using ELISA, the culture supernatants were screened for binding to recombinant human PDPN purified from LN229/hPDPN cells. After the limiting dilution, LpMab-10 (mouse IgG3, kappa) was established. LpMab-10 reacted with LN229/hPDPN, not with LN229, a PDPN-negative cell in Western blot analysis (Fig. 1A). LpMab-10 also recognized endogenous PDPN, which is expressed in a glioblastoma cell line LN319 and a lymphatic endothelial cell (LEC) in Western blot analysis (Fig. 1A) and flow cytometry (Fig. 1B). Although LpMab-10 did not react with a malignant mesothelioma cell line NCI-H226 and HEK-293T in Western blot analysis (Fig. 1A), it strongly recognized both cells in flow cytometry (Fig. 1B), indicating that LpMab-10 binds to native PDPN effectively. LN229 and CHO-K1, which do not express PDPN, were not detected by LpMab-10, indicating that LpMab-10 does not recognize non-related antigens in native condition (Fig. 1B). LpMab-10 also detects PDPN of a lung squamous cell carcinoma (SCC) cell line PC-10 and a normal mesothelial cell Met-5A (Fig. 1B), indicating that LpMab-10 is useful for detecting PDPN of both cancer cells and normal cells. We also performed Western blot analysis using LpMab-10 against several glycan-deficient PDPN transfectants, such as Lec1/hPDPN (N-glycan deficient), Lec2/hPDPN (sialic acid deficient), and Lec8/hPDPN (O-glycan deficient) (Fig. 1A). All transfectants of glycan-deficient PDPN were detected by LpMab-10, indicating that the epitope of LpMab-10 is independent of glycan. To investigate whether LpMab-10 reacts with synthetic peptides, we performed ELISA using several synthetic peptides of human PDPN (Fig. 1C). NZ-1 reacted with PLAG2/3 domain that corresponds to 38–51 amino acids of human PDPN (hpp38–51). In contrast, LpMab-10 strongly recognized PLAG1/2 domain that corresponds to 29–47 amino acids of human PDPN (hpp29–47). We next performed a kinetic analysis of the interaction of LpMab-10 with LN319 and PC-10 using flow cytometry. As shown in Figure 1D, KD was determined to be 8.755 × 10−9 M using LN319 and 9.830 × 10−9 M using PC-10, indicating that LpMab-10 possesses high affinity against human PDPN. We further performed antibody-overlay lectin microarray analysis of PDPN using LpMab-10 (Fig. 1E). PDPN strongly reacted with a α 2–3 sialic acid binder ACG, a sialic acid ± core1 binder Jacalin, and a sialo-mucin binder WGA, moderately reacted with a core1 ± sialic acid binder MPA, and faintly reacted with a poly LacNAc binder STL, indicating that LpMab-10 is useful in antibody-overlay lectin microarray analysis and PDPN possesses sialylated core1 structure.

Characterization of a novel anti-PDPN MAb, LpMab-10. (
Immunocytochemistry and immunohistochemistry
We next investigated whether LpMab-10 is useful for immunocytochemical and immunohistochemical analyses. LpMab-10 clearly stained the membrane of CHO/hPDPN, whereas it did not react with CHO-K1 cells (Fig. 2). LpMab-10 also stained endogenous PDPN expressed on LN319 cells, whereas it did not react with LN319/PDPN knock-out cells, indicating that LpMab-10 specifically recognizes human PDPN. As shown in Figure 3, LpMab-10 stained seminoma cells (Fig. 3C, D) and osteosarcoma cells (Fig. 3G, H) in a membranous/cytoplasmic staining pattern, indicating that LpMab-10 is very useful in immunohistochemical analysis.
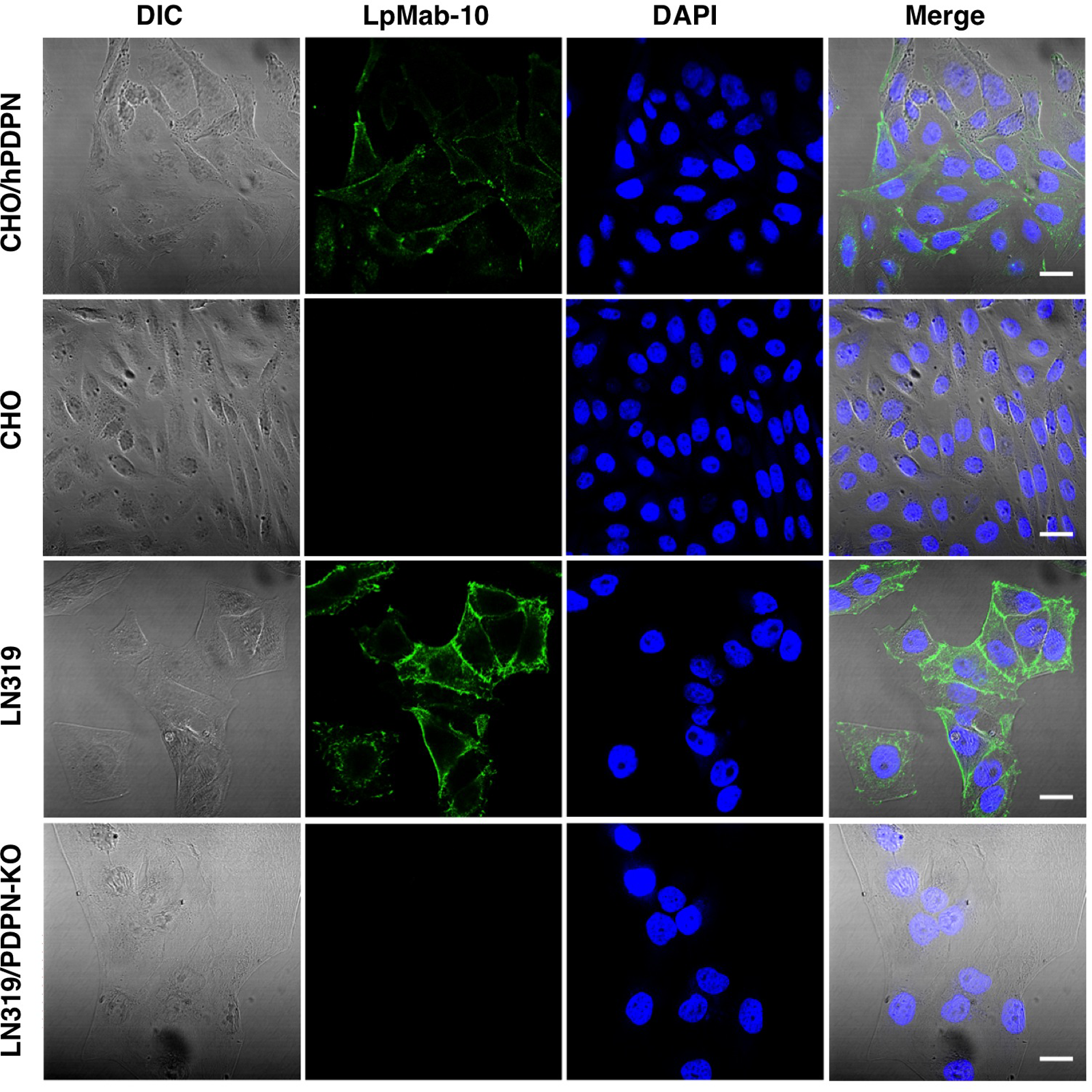
Immunocytochemical analysis. Cells were incubated with 1 μg/mL of LpMab-10 for 1 h at 4°C in a moist chamber. They were incubated with goat anti-mouse IgG-Alexa 488 for 30 min at room temperature. Cells were also treated with DAPI to stain the cell nuclei. Scale bars, 20 μm.
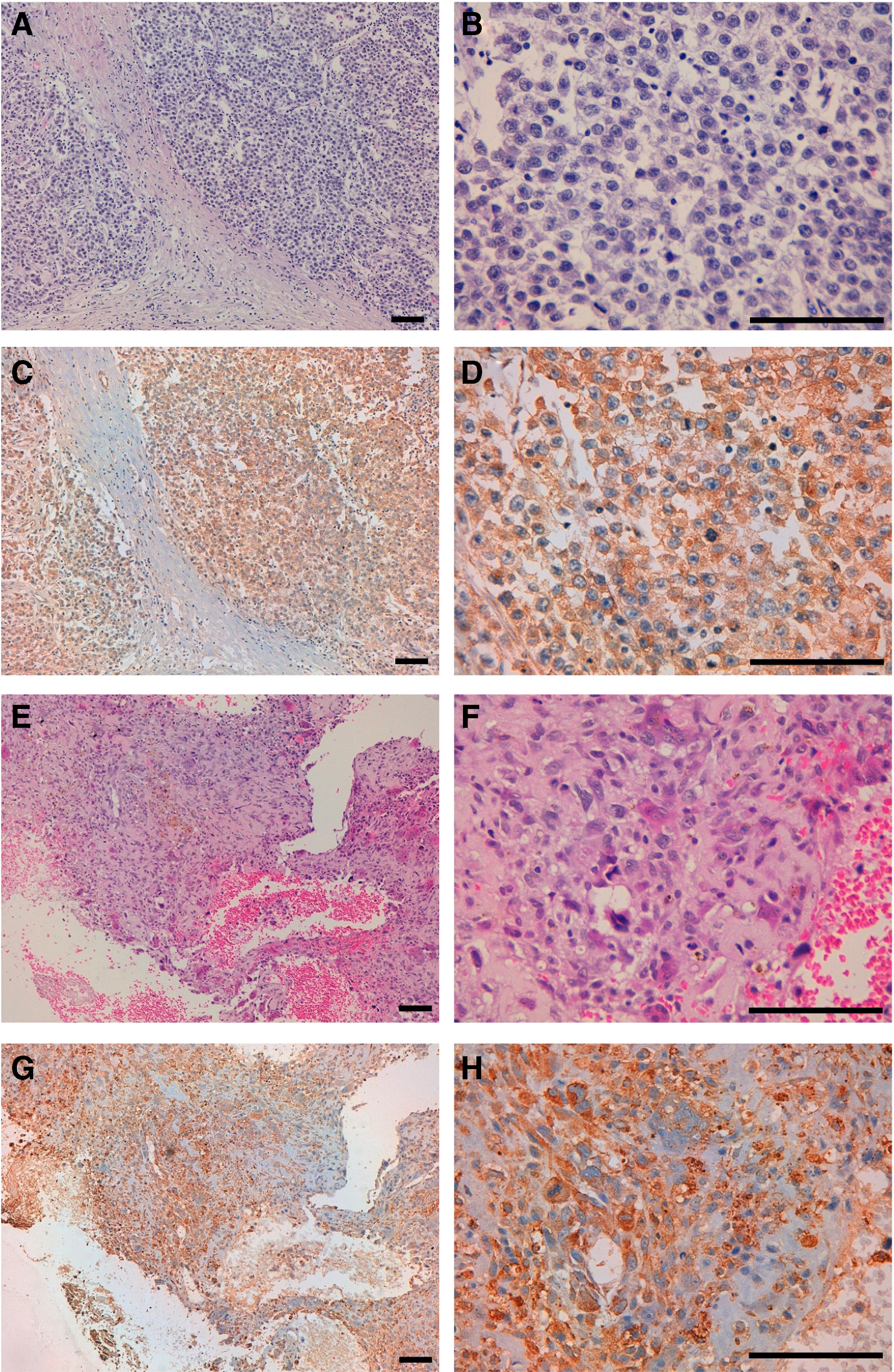
Immunohistochemical analysis by LpMab-10 against seminoma (
Epitope-mapping Western blot analysis and flow cytometry
The epitope of LpMab-10 was shown to be PLAG1/2 in ELISA (Fig. 1C); therefore, we next performed Western blot analysis to determine the LpMab-10 epitope in details. Western blot analysis showed that LpMab-10 reaction was lost in point mutations of 33–45 amino acids (Fig. 4A). Flow cytometric analysis revealed that LpMab-10 did not react with L37A and E38A; weakly recognized E33A, T34A, and P44A; moderately reacted with G39A, G40A, V41A, and G45A, indicating that Leu37 and Glu38 are much more important for LpMab-10 (Fig. 4B). Taken together, the PDPN epitope of LpMab-10 is Glu33-Gly45, and Leu37 and Glu38 are the most important amino acids.

Epitope mapping of LpMab-10 using flow cytometry and Western blot analysis. (
Discussion
Until now, ours and other groups have developed many anti-PDPN MAbs. To neutralize platelet aggregation activity by blocking the association between PDPN and CLEC-2, we produced NZ-1 MAb by immunizing rats with synthetic peptides of PLAG domain.(15) NZ-1 possesses very high binding affinity, which was clarified by several methods including ELISA, flow cytometry, Scatchard analysis, and BIAcore.(21,30) NZ-1 inhibited cancer-induced platelet aggregation and experimental metastasis by its neutralizing activity, indicating that NZ-1 is a candidate of anti-metastatic MAb.(15,29,31) In addition, NZ-1 was highly internalized into glioma cells and also accumulated efficiently into tumors in vivo.(21) NZ-1 and its rat-human chimeric anti-PDPN antibody (NZ-8) possess antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) against PDPN-expressing glioblastoma or malignant mesothelioma cells.(18,31) Therefore, NZ-1 is known to be an extremely useful anti-PDPN MAb. In contrast, rat MAbs are often avoided in immunohistochemistry, especially in pathological diagnosis, because almost all secondary antibodies or commercially available kits for immunohistochemistry have been developed against mouse MAbs or polyclonal antibodies, although many rat MAbs possess high-binding affinity and specificity. LpMab-10 shows the same reaction pattern with NZ-1 in Western blot analysis (Fig. 1A) and flow cytometry (Fig. 1B), demonstrating that LpMab-10 could possess more advantages compared with NZ-1 in several applications. Indeed, we used Dako Envision+ kit in immunohistochemistry, which is not applied for rat MAbs, indicating LpMab-10 is more advantageous than NZ-1.
Figure 4C shows that LpMab-10 epitope is independent of previously established anti-PDPN MAbs such as LpMab-2, LpMab-3, LpMab-7, LpMab-9, and NZ-1.(25,27,28,32,33) Furthermore, the minimum epitope of LpMab-10 is ETTGLEGGVAMPG, which consists of 13 amino acids, demonstrating that LpMab-10 epitope is longer than those of anti-PDPN MAbs. As we previously reported, Thr34 of mouse, rat, and hamster PDPNs might be O-glycosylated and shows the critical role for platelet aggregation.(34) Although we also demonstrated that Thr34 of human PDPN might not be glycosylated, and is not involved in platelet-aggregating activity,(35) the possibility of glycosylation of Thr34 was suggested in platelet aggregation and in a tumor metastasis model in another study.(22) It was also suggested that O-glycosylation sites are predicted by the NetOGlyc prediction tool, and Thr32, Thr34, and Thr35 of PLAG1 might be O-glycosylated.(36) These studies did not investigate the O-glycosylation directly because there was no tool to detect the O-glycosylation of each Thr. As shown in Figure 4C, we have produced several site-specific MAbs against each O-glycosylated Ser/Thr of human PDPN; therefore, we can directly detect O-glycosylation of Thr25, Thr34, Thr52, Thr55, Ser56, and Thr76. Because Thr34 of PLAG1 is critical for LpMab-10 recognition (Fig. 4A, B), and O-glycan is not included in LpMab-10 epitope (Fig. 1A), it is more likely that Thr34 of PLAG1 is not O-glycosylated.
In conclusion, LpMab-10 could be advantageous for investigating expression and function of PDPN in cancers and normal tissues. By using MAbs against different Ser/Thr-including epitopes of human PDPN, we can determine whether Ser/Thr residues of PDPN are O-glycosylated. Further, different epitope-possessing anti-PDPN MAbs should be established as powerful tools for investigating function of PDPN in cancers and normal tissues in the future.
Footnotes
Acknowledgments
We thank Noriko Saidoh and Kanae Yoshida for their excellent technical assistance. This work was supported in part by the Platform for Drug Discovery, Informatics, and Structural Life Science (PDIS) from Japan Agency for Medical Research and Development from AMED (Y.K.); by the Basic Science and Platform Technology Program for Innovative Biological Medicine from AMED (Y.K.); by the Regional Innovation Strategy Support Program from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (Y.K.); and by JSPS KAKENI Grant Number 25462242 (Y.K.) and Grant Number 26440019 (M.K.K.).
Author Disclosure Statement
The authors have no financial interests to disclose.