
Abstract
A major clinical problem in the treatment of breast cancer is mortality due to metastasis. Understanding the molecular mechanisms associated with metastasis should aid in designing new therapeutic approaches for breast cancer. Trastuzumab is the main therapeutic option for HER2+ breast cancer patients; however, the molecular basis for trastuzumab resistance (TZR) and subsequent metastasis is not known. Earlier, we found expression of basal-like molecular markers in TZR tissues from patients with invasive breast cancer.(1) The basal-like phenotype is a particularly aggressive form of breast cancer. This observation suggests that TZR might contribute to an aggressive phenotype. To understand if resistance to TZR can lead to basal-like phenotype, we generated a trastuzumab-resistant human breast cancer cell line (BT-474-R) that maintained human epidermal growth factor receptor 2 (HER2) overexpression and HER2 mediated signaling. Analysis showed that nuclear factor-kappa B (NF-κB) was constitutively activated in the BT-474-R cells, a feature similar to the basal-like tumor phenotype. Pharmacologic inhibition of NF-κB improved sensitivity of BT-474-R cells to trastuzumab. Interestingly, activation of HER2 independent NF-κB is not shown in luminal B breast cancer cells. Our study suggests that by activating the NF-κB pathway, luminal B cells may acquire a HER2+ basal-like phenotype in which NF-κB is constitutively activated; this notion is consistent with the recently proposed “progression through grade” or “evolution of resistance” hypothesis. Furthermore, we identified IKK-α/IKK-β and nuclear accumulation of RelA/p65 as the major determinants in the resistant cells. Thus our study additionally suggests that the nuclear accumulation of p65 may be a useful marker for identifying metastasis-initiating tumor cells and targeting RelA/p65 may limit metastasis of breast and other cancers associated with NF-κB activation.
Introduction
B
Overexpression of the human epidermal growth factor receptor 2 (HER2) occurs in approximately 20–25% of human breast cancers found on luminal A and luminal B breast cancers and is an indicator of poor prognosis for these subtypes.(5) A recombinant humanized anti-HER2 monoclonal antibody, trastuzumab (Herceptin), is approved for the treatment of HER2-overexpressing breast cancer and is effective in patients with HER2+ breast cancer. However, despite the clinical benefits of these HER2-targeted therapies, almost 50% of patients with HER2+ breast cancers fail to respond to trastuzumab and the vast majority of tumors that respond to trastuzumab develop resistance within 1–2 years of treatment.(6)
In some cases, the combination of trastuzumab with chemotherapy treatment improved response rates and increased overall survival rates compared to chemotherapy alone.(7–9) Unfortunately, as is the case for trastuzumab monotherapy, many patients treated with trastuzumab plus chemotherapy develop progressive disease within one year.(7–9) In particular, breast cancer patients with HER2-overexpressing, luminal B and some basal-like breast cancer subtypes had poor prognosis post adjuvant therapy.(10) Luminal A patients display a short-term risk of relapse, but after 3 years remain stable. For luminal B patients, on the other hand, the risk of relapse occurs during the first 5 years and recurrence happens nearly 20 months post-surgery.(11) Thus, acquired resistance to trastuzumab remains an important issue in the clinical treatment of HER2+ breast cancer.
These observations suggest that there is an immediate need to address trastuzumab resistance (TZR) in patients with the relapsing luminal B breast cancer subtype and prevent metastasis-initiating tumor cells. Understanding the molecular mechanisms that contribute to the acquired resistance will ultimately allow for the identification of biomarkers that can be used to predict response to trastuzumab therapy and prevent metastasis, as well as identification of new molecular targets for the development new therapeutics.
Current therapy for HER2+ breast cancer is directed at the ectodomain of the HER2 receptor.(12,13) In metastatic breast cancer, a combination treatment of trastuzumab and chemotherapeutics prolongs survival.(9) However, both de novo and acquired resistance to trastuzumab is prevalent and overall survival gains have yet to be realized with trastuzumab monotherapy. Therefore, more effective combinations containing trastuzumab are sought for HER2-expressing breast cancer.
In addition to HER2-mediated signaling, increasing evidence suggests that other pathways, such as activation of nuclear factor-kappa B (NF-κB) and STAT, are key molecular events in driving malignancies by dysregulating apoptotic, inflammatory, and immune responses.(14) Elevated levels of NF-κB are frequently detected in many diseases, including breast cancer.(15–17) The activation of NF-κB in human breast cancer is confined predominantly to the estrogen receptor (ER)-negative subtype of cancers, particularly those that express members of the EGF family of receptors, including the EGF receptor (ErbB1) and ErbB2 (HER2/neu).(15,18) This trend was confirmed in tissue samples from patients with breast cancer.(15) Growth factor receptors are known to activate NF-κB pathways following treatment of ErbB2-expressing breast cancer cells with either EGF or heregulin β1.(12,13,15,18) However, the functions of NF-κB in TZR breast cancers prior to exposure to cytotoxic agents are unknown.
Numerous studies suggest that several mechanisms mediate resistance to trastuzumab therapy: expression of truncated HER2 receptor (p95)(19) and epitope masking by co-receptors such as CD44, MUC4, and hylauronan.(20) However, the evolution of TZR is not fully understood.
In an earlier study, we analyzed basal-like molecular markers in TZR patient tissues and HER2+ breast cancer cells and found that the expression of EGFR and constitutively activated Akt kinase pathways in HER2+ “basal-like” breast cancer correlated with poor prognosis.(1) These studies showed that HER2+ tumor cells also share some molecular features of basal-like or triple-negative breast cancer cells. Since NF-κB is constitutively activated in basal-like tumor cells, we hypothesized that chronic treatment of HER2+ breast cancers may activate NF-κB to acquire basal-like phenotype and exhibit growth and metastasis.
In this study, to understand the changes in the molecular profile due to chronic treatment of trastuzumab, we established a clone derived from the HER2-overexpressing BT-474 breast cancer cell line (BT-474-R) that acquired trastuzumab resistance (TZR). Although the activation of NF-κB is reported to be confined predominately to the ER-negative subtype of cancers, we found constitutive activation of NF-κB in the trastuzumab-resistant HER2-overexpressing BT-474-R breast cancer cell line with accumulation of IKKs and nuclear-RelA/p65. We suggest that nuclear RelA and p65 may serve as molecular markers of metastasis-initiating cells and perhaps serve as prognostic indicators following current adjuvant therapies. Additionally, agents that can block NF-κB-activating kinases (IKKs) may also offer new clinical therapeutic value for trastuzumab-resistant breast cancer and prevent metastasis.
Materials and Methods
Establishing trastuzumab-resistant breast cancer cell line
BT-474 breast cancer cells were chronically exposed to increasing concentrations of trastuzumab (5, 10, 20, 40 μg/mL) in vitro for a total of 10 months. The resistant cells (i.e., displayed the same growth rate as that of parental cells) were selected and grown in Dulbecco's modified Eagle's medium (DMEM, Mediatech, Manassas, VA). The resistant cells were named BT-474-R.
Cell culture
Human breast cancer cell lines BT-474 and BT-474-R were grown in DMEM with 10% fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin. Both cell lines were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Quantitative PCR and RT-PCR
Total RNA was extracted using TRIsure (Bioline, Taunton, MA) according to standard guanidium-phenol-chloroform extraction procedures. The amount and integrity of the RNA were assessed by measurement of absorbance at 260 and 280 nm using an Infinite 200 PRO (Tecan Group, Mannedorf, Switzerland). Total RNA was reverse transcribed into first strand cDNA using iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. Quantitative real-time PCR was performed using SsoFast EvaGreen (Bio-Rad Laboratories) according to the manufacturer's protocol. PCR was performed at 98°C for 2 min followed by 44 cycles at 98°C for 2 s, 60°C for 5 s, and the dissociation step at 65°C for 5 s, 95°C for 5 min. A comparative cycle of threshold fluorescence (CT) value was normalized to the CT value of 18S ribosomal 1 (RN18S1) rRNA in the same sample. The relative expression changes of mRNA in BT-474-R cells were calculated using ΔΔ Ct method with BT-474 as reference.
Western blot analysis
For total protein extraction, cells were lysed in RIPA lysis buffer: 50 mM Tris-HCl (pH 8.0), 150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS, Sigma-Aldrich, St. Louis, MO), containing protease inhibitor cocktail (Sigma-Aldrich), and phosphatase inhibitor cocktail 2 (Sigma-Aldrich). For the separation of nuclear protein and cytoplasmic protein, cells were lysed in NE-PER Nuclear and Cytoplasmic Extraction reagents (Thermo Scientific, Brookfield, WI) according to the manufacturer's instructions. Lysates were centrifuged at 15,000 g for 15 min at 4°C and the protein concentration was determined by DC protein assay reagent (Bio-Rad Laboratories). Western blot analysis was performed according to the guidelines of Trans-Blot Turbo Transfer system protocol (Bio-Rad Laboratories). In brief, 30–50 μg of total proteins were heated for 5 min at 95°C, then separated on a 4–20% SDS-polyacrylamide gel and electrotransferred to a nitrocellulose membrane (Bio-Rad Laboratories). β-actin was used as loading control (data not shown). A standard enhanced chemiluminescence (ECL) detection system was used to detect the bands. Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline-Tween-20 (TBS-T) buffer and incubated overnight at 4°C with the following primary antibodies: 1:250 rabbit polyclonal anti-HER2 (sc-284, Santa Cruz Biotechnology, Santa Cruz, CA), 1:250 rabbit polyclonal anti-phospho-HER2 (sc-1235R, Santa Cruz Biotechnology), 1:250 rabbit polyclonal anti-p65 (sc-109, Santa Cruz Biotechnology), 1:1000 rabbit polyclonal anti-Akt (#9272, Cell Signaling Technology, Danvers, MA), 1:1000 rabbit polyclonal anti-phospho-Akt (#9271, Cell Signaling Technology), 1:250 rabbit polyclonal anti-IκBα (sc-847, Santa Cruz Biotechnology), 1:250 rabbit polyclonal anti-phospho- IκBα (sc-8404, Santa Cruz Biotechnology), 1:1000 of rabbit IKKα or IKKβ antibody (Cell Signaling Technology), or 1:1000 mouse monoclonal anti-beta actin (ab8226, Abcam, Cambridge, MA). After washing with TBS-T, membranes were incubated with peroxidase-conjugated secondary antibody in TBS-T for 1 h (1:5000 dilution) at room temperature. Blots were washed and hybridization signals were measured by ECL detection system using Immun-Star WesternC (Bio-Rad Laboratories) and ChemiDoc XRS System (Bio-Rad Laboratories). Images were analyzed quantitatively using ImageJ 1.46r image analysis software.
Cell survival assay
Cell survival was quantitated using a colorimetric MTT assay according to the procedures described previously.(21) 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich. 100 μL of target cell suspension (1 × 104 cells) were added to each well of a 96-well plate, and the plate was incubated for 24 h at 37°C in a humidified 5% CO2 atmosphere. Cells were incubated for another 72 h with or without trastuzumab, BAY11-7082, or both. After drug treatment, 100 μL of 10 μM MTT working solution were placed in each well and the plates were incubated for 3 h at 37°C. After addition of solubilizing solution, the absorbance values were measured with a microplate reader at 570 nm. The percentage of survival was calculated using the following formula: survival percentage = (absorbance of drug treated wells – blank wells) / (absorbance of untreated wells – blank wells) × 100.
Soft agar colony-forming assay
Base layers consisting of growth medium containing 0.5% low-melting point agarose (Life Technologies, Grand Island, NY) were poured onto 6-well plates and allowed to solidify. Cells (1 × 104 per well) were plated in triplicate in top layers consisting of growth medium containing 0.3% agarose. Cells were stained 1–2 week(s) later with 0.2% iodonitrotetrazolium chloride (Sigma-Aldrich) and colonies composed of more than or equal to 50 cells were counted manually in five randomly selected fields.
Flow cytometry
Flow cytometer LSRII (BD Biosciences, San Jose, CA) was used to quantitate expression levels of proteins labeled by fluorescent antibodies. Analysis of flow cytometry data was carried out with FlowJo software (v. X, Tree Star, Ashland, OR). 1 × 105 cells were collected in a FACS tube, centrifuged, and suspended in 25 μL of FACS buffer (5% FBS in PBS) and then 10 μL of phycoerythrin (PE)-conjugated mouse monoclonal anti-human ErbB2 antibody (FAB1129P, R&D Systems, Minneapolis, MN) was added. After 30 min of incubation on ice, cells were washed by 5X diluted FACS buffer and resuspended with 500 μL of 1X binding buffer. The cells were then scanned for fluorescence intensity in FL-2 and analyzed by flow cytometry.
Statistical analysis
Experiments were repeated independently a minimum of three times and values were expressed as means ± standard deviation (SD). Differences were assessed by one-way ANOVA followed by Scheffe's F-test. A value of p < 0.05 (or p < 0.01 where noted) was used to indicate statistical significance.
Results
Validation of trastuzumab-resistant breast cancer cells
Resistance to trastuzumab has been extensively studied; these studies revealed antibody (epitope) masking, loss of epitope, and activation of survival pathways including PI3K/Akt pathways.(19,22–25) To rule out loss of epitope or epitope-masking effects, first, we validated the overexpression of HER2 receptor and function in BT-474 cells. The HER2 expression remained unaltered between the parental cells (BT-474) and the resistant cells (BT-474-R), both at the mRNA (Fig. 1A) and protein levels (Fig. 1B). Western blot analysis with anti-phospho-HER2 antibody demonstrated that the HER2 protein was constitutively active in both cell lines and that the level of phosphorylation was not altered between these two lines (Fig. 1B). Moreover, the cell surface expression of HER2 is similar in both cell lines as determined by flow cytometry (Fig. 1C). Finally, the colony formation assays showed that the transformation potential of BT-474-R is significantly higher than that of the parental cells (Fig. 1E, F). These results suggest that the two cells are similar, but phenotypically different.

Validation of trastuzumab-resistant HER2+ breast cancer cell line (BT-474-R). (
To determine the status of HER2 signaling, the cells were treated with trastuzumab for 72 h, and cell survival was measured by MTT assay. As shown in Figure 1D, BT-474-R cells showed significantly more resistance to trastuzumab than did parental cells. At the highest dose of 2100 μg/mL of trastuzumab, about 77% of the resistant cells remained viable, while only about 22% of the parental cells survived (Fig. 1E). On the other hand, both cell lines were sensitive to lapatinib, a dual receptor tyrosine kinase inhibitor, in a dose-dependent manner at a concentration range of 0.01–50 μM (Suppl. Fig. 1). These observations suggest that the HER2 signaling is active, but may impart resistance to trastuzumab by co-opting other cell surface receptors, as suggested previously.(20) Altogether, these results suggest that TZR is not due to either enrichment of HER2 on cell surface or epitope masking.
NF-κB is constitutively activated in BT-474-R
Although NF-κB is known to be activated in basal-like(26) and drug-resistant HER2+ tumor cells,(27) it is not known whether NF-κB is activated in TZR HER2+ tumor cells.(28,29) The fact that HER2 expression is unaltered between BT-474 and BT-474-R suggests that there might be alternate pathways. Since NF-κB–mediated inflammation is one of the key factors in the promotion of survival genes, we first examined the mRNA expression of NF-κB family members in both cell lines. Although mRNA expression of NFKB1 (p50) and NFKB3 (p65) was the same in both cell types, NFKB2 (p52) and expression of RELB was decreased (Fig. 2A). To compare the constitutive activation of NF-κB in these cells, we examined nuclear localization of p65 by Western blot (Fig. 2B). Quantification of nuclear p65 protein levels indicated a 4.1-fold increase in BT-474-R cells than in parental cells. These results suggest that NF-κB is constitutively activated in the trastuzumab-resistant cells (Fig. 2B).
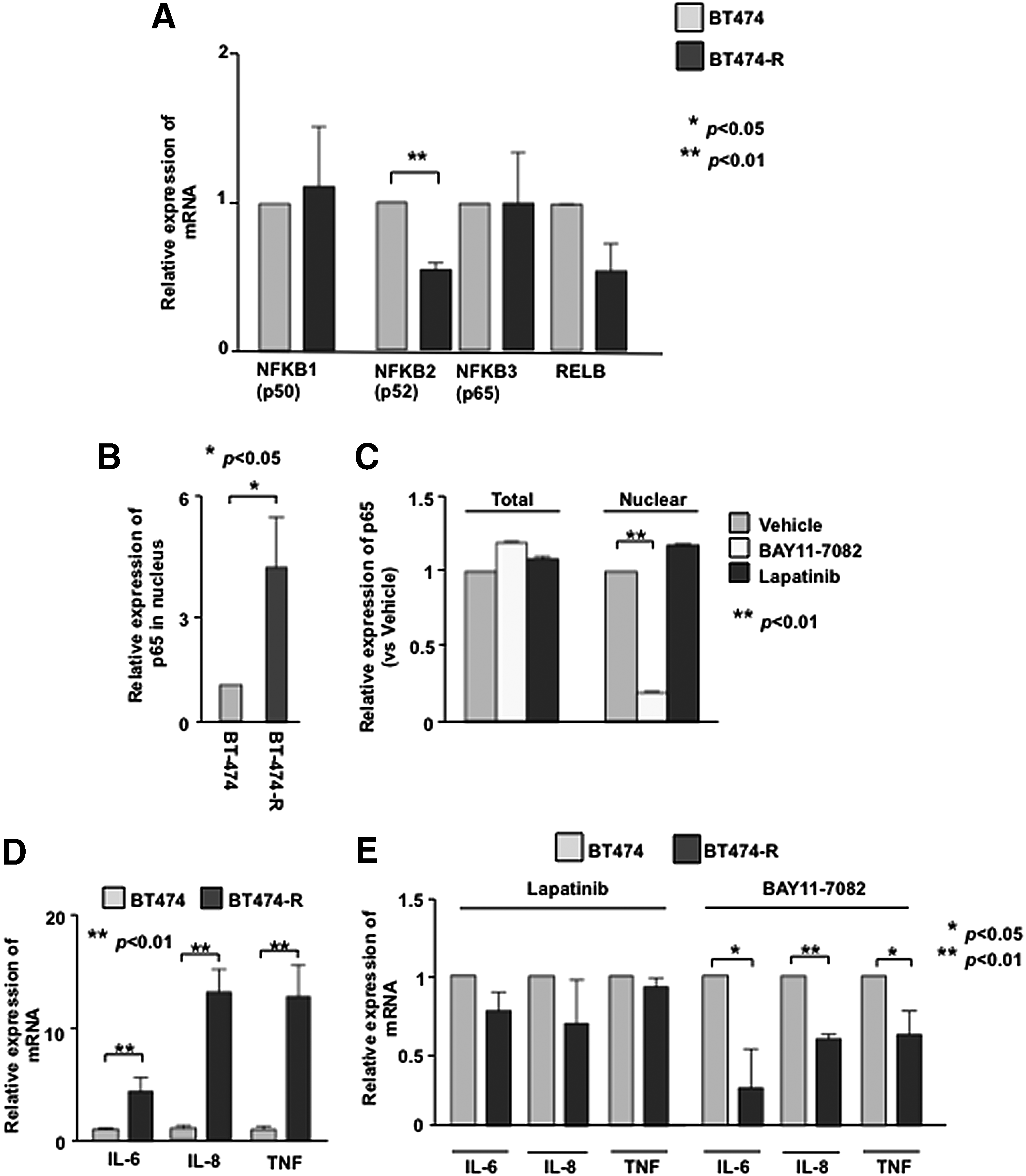
NF-κB is activated in trastuzumab resistant cell. (
To assess whether NF-κB activation co-opted HER2 signals, we analyzed NF−κB activation and HER2 signal by using the NF-κB inhibitor, BAY 11-7082, or lapatinib in BT-474-R cells. The resistant cells treated with BAY 11-7082 significantly decreased the level of p65 in the nucleus (Fig. 2C). Lapatinib alone did not have any effect on the nuclear localization of p65 (Fig. 2C), suggesting that NF-κB signal may be decoupled from HER2 signaling and independently activated to augment TZR.
To examine whether NF-κB activates pro-inflammatory signals, we also examined the expression of a subset of pro-inflammatory cytokines, IL-6, IL-8, and TNF in both cell lines. Overexpression of HER2 has been shown to secrete IL6 in TZR cells.(28,29) At the basal level, expression of these pro-inflammatory cytokines was significantly up-regulated in the resistant cells with a 4.4-fold, 13.2-fold, and 12.9-fold up-regulation (respectively) relative to BT-474 cells (Fig. 2D). As expected, NF-κB inhibitor suppressed the induction of pro-inflammatory cytokines in resistant cells (Fig. 2E). Collectively, these results indicate that NF-κB is constitutively activated in resistant cells and transcriptionally active to induce pro-inflammatory cytokines as a survival mechanism.
Development of trastuzumab resistance depends on NF-κB
We examined the dose response of BT-474 and BT-474-R cells to NF-κB inhibitor BAY 11-7082, both alone and in combination with trastuzumab. BT-474-R cells showed significantly higher sensitivity to BAY 11-7082 versus BT-474 cells (Suppl. Fig. 2). At 10 μM of BAY 11-7082, more than 90% of parental BT-474 cells remained viable while only 67% of BT-474-R cells were viable (Fig. 3A). Combination treatment (10 μM BAY 11-7082 + 21 μg/mL trastuzumab) resulted in significant inhibition of viability of BT-474-R cells (52% reduction) (Fig. 3A). A similar observation was also noted in the colony formation assay; treatment with either BAY 11-7082 alone or in combination with trastuzumab enhanced anti-tumor effects on BT-474-R cells (Fig. 3B–D). Taken together, these data indicate that NF-κB plays a dominant role in the development of TZR in the BT-474 cell line.
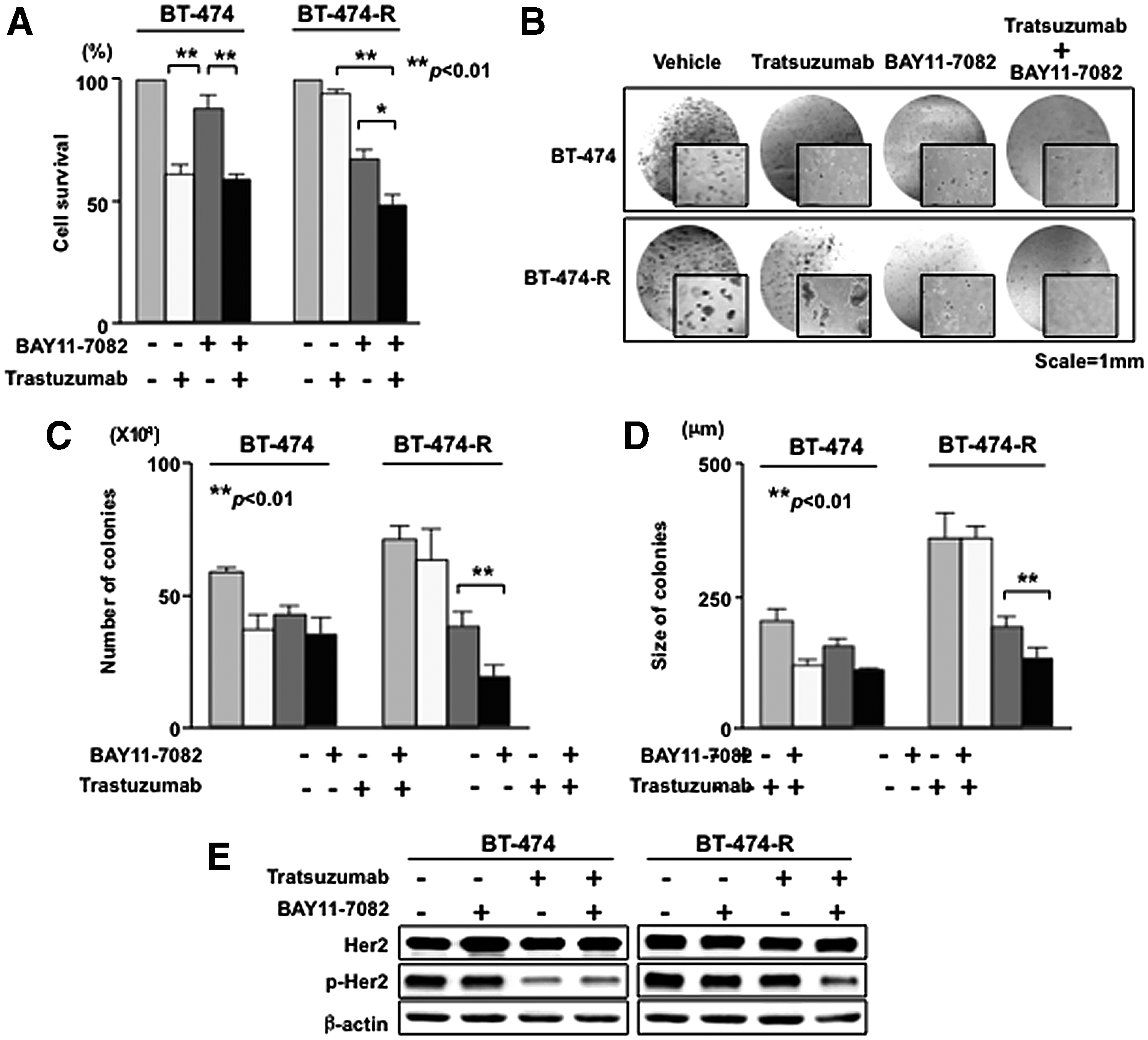
NF-κB inhibitor reduces cell growth and recovers sensitivity to trastuzumab in resistant cells. (
Next, we tested whether BAY 11-7082 enhances the effect of trastuzumab on the inhibition of HER2 activation alone or in combination with trastuzumab. In parental (BT-474) cells, the phosphorylation of HER2 is significantly reduced in the presence of trastuzumab alone and in the presence of both trastuzumab and BAY 11-7082 (Fig. 3E, left panel). Treatment by trastuzumab alone had no significant effect on the phospho-HER2 in the resistant (BT-474-R) cells (Fig. 3E, right panel); however, phosphorylation of HER2 was significantly reduced when resistant cells were treated with both drugs (Fig. 3E, right panel). These data suggest that downstream NF-κB and HER2 signals are co-opted in the resistant cells.
Mechanism of NF-κB activation in trastuzumab-resistant cells
The NF-κB inhibitory protein IκB controls NF-κB activation.(30,31) Following stimulation with activators, the inhibitor of κB kinases, IKK-α, and IKK-β phosphorylates IκB, which leads to IκB degradation facilitating p65-p50 nuclear translocation for inflammatory and survival gene activation. Here, we first tested whether there was any change in IκB expression or activation in resistant cells compared to BT-474 cells. As indicated in Figure 4A, there was no change in the expression of IκB in either cell line and the phosphorylation of IκB in BT-474-R cells was sensitive to NF-κB, but not to lapatinib treatment as expected (Fig. 4B). Next, we investigated whether there was any change in the expression level of IKK kinases, which control IκB degradation. Interestingly, we found significant up-regulation of both IKK-α and IKK-β kinases only in the BT-474-R cells (Fig. 4C), which indicates that NF-κB activation occurs primarily through the IKK complex.
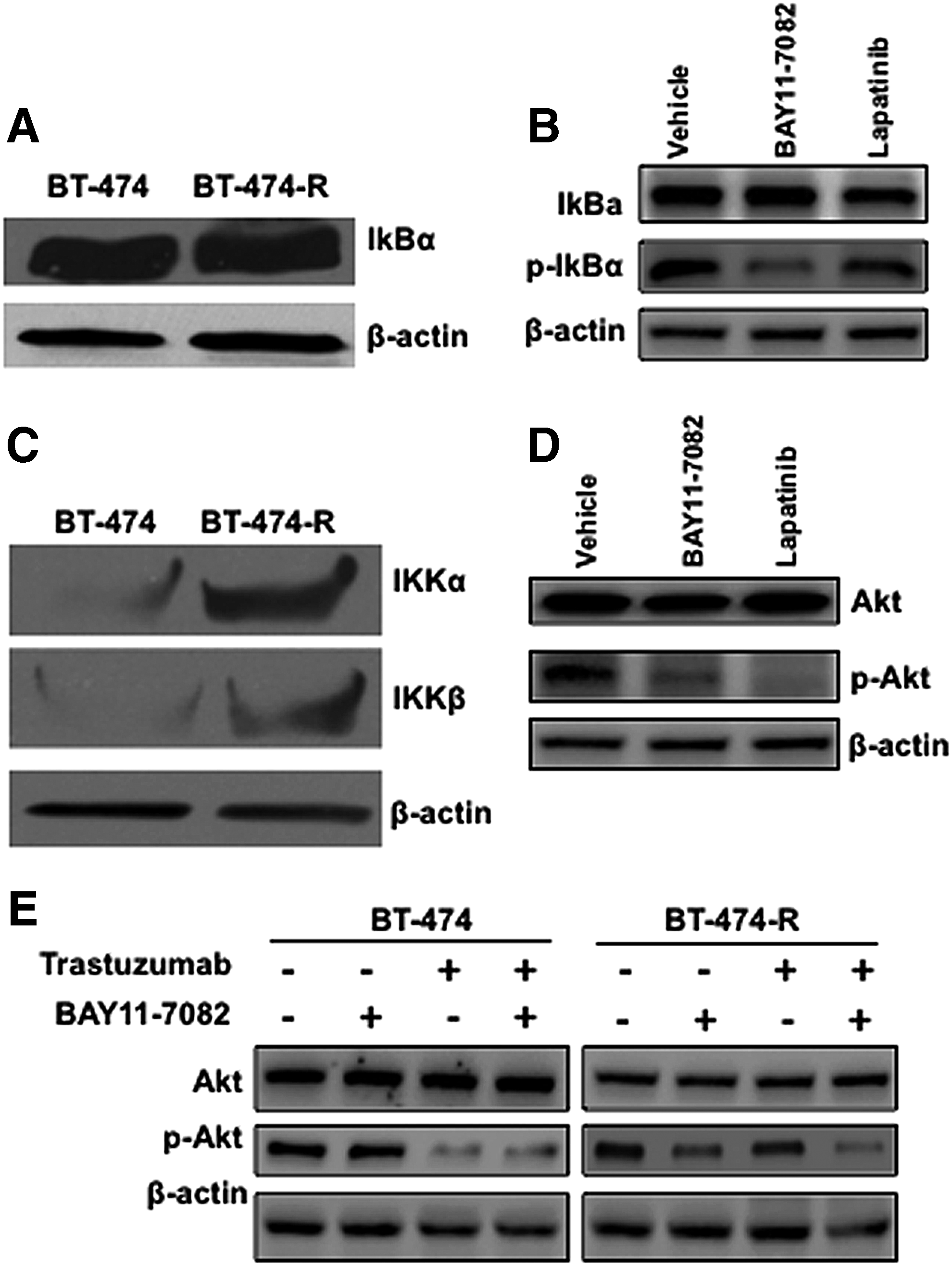
Possible mechanism of NF-κB activation in resistant cells. (
HER2 is known to activate both PI3/Akt and NF-κB pathways in chemotherapy and radiation-resistant breast cancer cells.(32–34) Therefore, we tested whether the NF-κB activation downstream of HER2 in resistant cells depends on the PI3/Akt pathway. In the BT-474-R, treatment with both BAY 11-7082 and lapatinib inhibits the Akt activity significantly, as indicated by the phospho Akt blot (Fig. 4D), without affecting Akt expression. On the other hand, in the parental BT-474 cells, the phosphorylation of Akt was decreased in the presence of the HER2 kinase inhibitor but not in the presence of NF-κB inhibitor (Fig. 4E). Interestingly, Akt activation is significantly affected by the NF-κB inhibitor and partially affected by the HER2 inhibitor in resistant cells (Fig. 4E). When treated together, the PI3K/Akt signal is significantly affected in both cells. Our data thus indicate that NF-κB activation in resistant cells could be initiated by both IKK kinases through the canonical pathway and the integrated signal from the HER2 activation pathway.
Discussion
Invasive breast carcinomas are a heterogeneous group of tumors with different biologic behaviors, prognoses, and responses to therapy. Two major subgroups in breast cancer are based on the status of estrogen receptor (ER) and HER2 expression. Within the ER subgroup, there are five distinct subtypes of breast carcinomas: (a) luminal A; (b) luminal B, largely associated with ER+; and three subtypes associated with low ER status: (c) basal-like; (d) HER2 overexpressing; and (e) normal-like breast cancer.(35) While subtype luminal A carries a good prognosis, luminal B breast cancer subtype has a high chance of recurrence post-adjuvant therapy and has poor survival rates due to metastasis. The main targeted therapy for luminal B breast cancer patients is trastuzumab. Our observations of breast tissues from patients who manifested resistance to trastuzumab suggest that HER2+ tumor cells exhibit basal-like molecular markers.(1) The present study collectively indicates that HER2+ luminal B breast cancer cells with acquired resistance to prolonged use of trastuzumab alone show a constitutive activation of NF-κB decoupled from HER2 signaling as a survival mechanism. An elevated level of activated NF-κB is detected in human breast cancer cells and tumor tissue specimens predominantly in HER2-overexpressing ER-negative breast(15,16,36) and in chemoradiation resistant breast cancers.(32,37) As a novel finding, our data suggest that NF-κB activation is correlated with trastuzumab resistance not only in ER-negative breast cancer, but also in ER-positive luminal B breast cancer, which is prone to relapse.(11)
Investigations related to the molecular mechanisms of acquired and inherent trastuzumab resistance reveal that several pathways can lead to TZR breast cancer. In the case of inherent resistance trastuzumab, loss of HER2 receptors or epitope masking due to large MUC4 receptors or activation of PI3K/Akt pathways has been attributed in basal-like HER2+ cells such as JIMT-1(38,39) and HCC-1954.(1,40) In the case of acquired resistance (as in the case of our study), we found that HER2 expression and phosphorylation are not altered in trastuzumab-resistant BT-474-R cells developed by a long-term (∼10 months) treatment with the drug. Others have examined trastuzumab resistance in vitro using resistant clones. Dr. Arteaga's group has studied TZR by continuous growth of BT-474 cells in athymic nude mice under long treatment. Their results indicate that the resistant clones have not lost HER2 expression, but overexpress epidermal growth factor receptor (EGFR) and HER3.(41,42) On the other hand, Kute and colleagues(23) developed resistant cells by treating with the antibody over 2 weeks and found that PI3K and overexpression of EGFR mediate resistance. Chan and colleagues(43) cultured cells for 6 months and concluded that p27 expression is an indicator of TZR. The resistant cell lines developed in this study represent an intermediate period from other reports (2 weeks to 2 year cultures) and thus provide a snapshot of the evolution of TZR (Fig. 5).
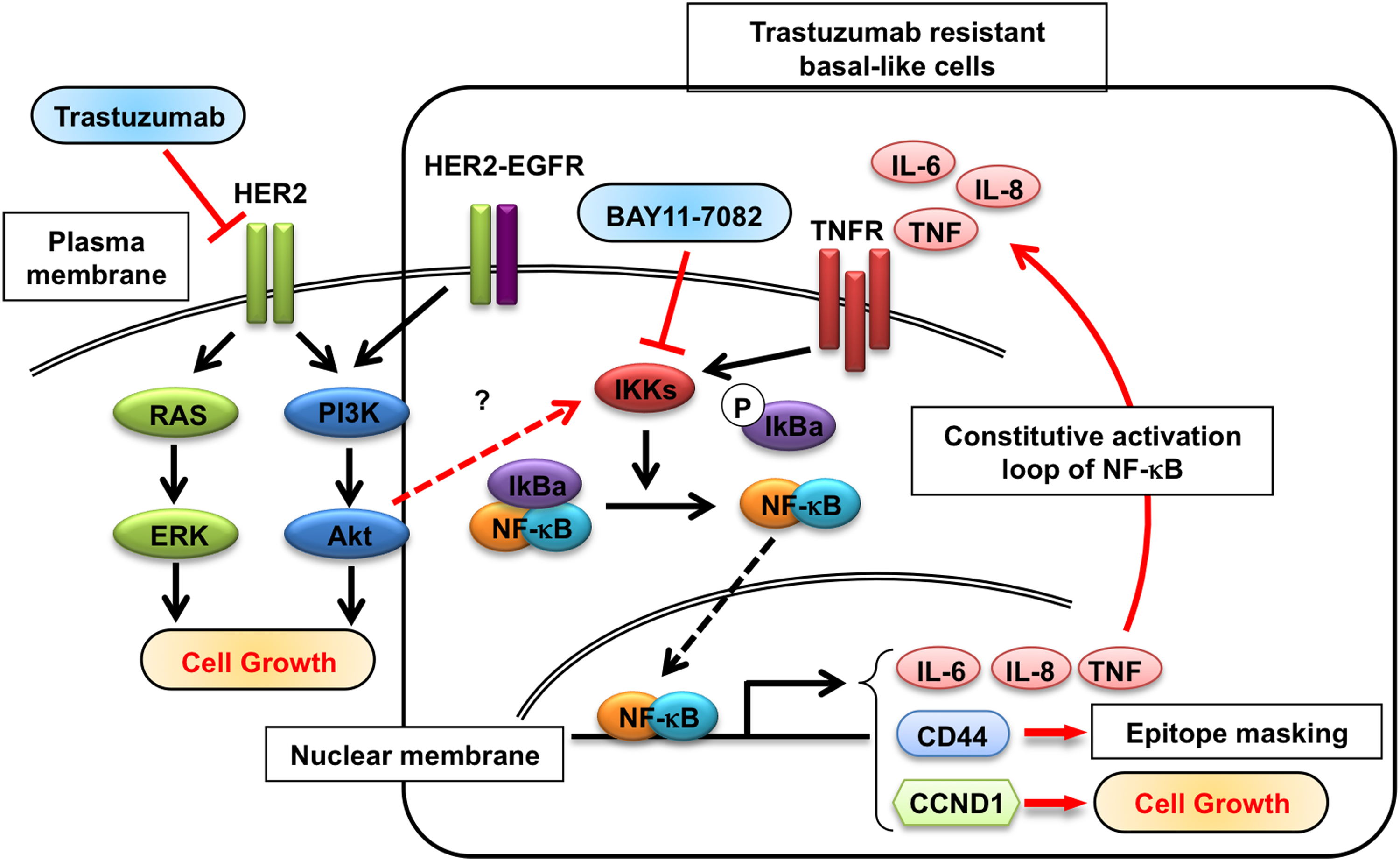
Model for linkage of HER2 and NF-κB signals in the evolution of trastuzumab resistance in HER2+ breast cancer cell line. In trastuzumab-resistant BT-474 cells, but not in sensitive cells, constitutively active NF-κB leads to nuclear accumulation of RelA/p65, which in turn can activate expression of target pro-inflammatory genes (such as IL-6, IL-8, and TNFα) other survival genes including CD44 and cell cycle regulators. Pro-inflammatory cytokine TNFα sustains the active state of NF-κB through a positive feedback, leading to constitutive activation of autocrine NF-κB target gene expression. Some of the pathways implicated are inferred from other studies and are depicted to illustrate a global effect following NF-κB activation that can ensue during TZR evolution. A black dotted arrow indicates nuclear translocation of p65/p50. Other uncharacterized pathways under investigation are shown in red dotted arrows.
Modulation of apoptotic regulators, including NF-κB, as a potential strategy for treating breast cancer is supported by a number of studies.(44) NF-κB activation in breast cancer cells has been shown to increase trastuzumab resistance,(45) supporting the idea of NF-κB as a potential target in trastuzumab-resistant breast cancers. Our results of significant nuclear accumulation of p65 (RelA), the most predominant active complex of NF-κB family in epithelial cells in association with p50 subunit, indicate that NF-κB pathway is functional in resistant cells. This is also supported by our observation that this accumulation of p65 in resistant cells is sensitive to NF-κB inhibitor BAY11-7082. This p50/RelA NF-κB dimmer complex is traditionally considered the signal output of the canonical NF-κB pathway. Our data also suggest that pro-inflammatory cytokines in resistant cells may contribute to establishing constitutive activation of NF-κB. Nuclear accumulation of NF-κB activates transcription of a number of genes whose promoters contain NF-κB response elements, including pro-inflammatory cytokines (IL-6, IL-8, and TNF). Gene expression analysis of p65 knockdown cells led to a decrease in gene transcription levels of IL-6, IL-8, and TNF-α.(46) These data are consistent with our report as we observe the induction of all these cytokines and chemokines in addition to the accumulation of p65 in trastuzumab-resistant cells. Moreover, our study also indicates that this induction of chemokine and cytokine levels is significantly sensitive to NF-κB inhibitor treatment, although there is marginal effect after lapatinib treatment.(47,48)
Activation of NF-κB signal follows both canonical and non-canonical pathways, and crosstalk between these two pathways appears to exist.(49) Our experiments indicated that the level of IκB is unaltered in both cell lines and the phosphorylation of IκB in resistant cells is sensitive to NF-κB inhibitor treatment. Among the IKKs, IKKβ is the major kinase in phosphorylating and degrading IκB in canonical pathways.(50) IKKα is involved in both canonical and non-canonical pathways.(46,51,52) Our data indicate that the canonical pathways, involving both IKKα and IKKβ, are important for the activation of NF-κB in trastuzumab-resistant BT-474-R cells. Silencing and overexpression of both IKK kinases will determine the role of this kinase individually in the development of resistance in the BT-474-R cell line. Experiments are underway to address these questions. Moreover, the up-regulation of the HER2 downstream signal pathway is another cause of trastuzumab resistance. PTEN loss(36,53) has been implicated in trastuzumab resistance by increasing PI3K/Akt(33,45) and activating IL-6 to mediate cancer stem cells.(29) Our data have established that the activation of PI3K/Akt pathway is sensitive to the NF-κB inhibitor in resistant cells. This confirms that the PI3K/Akt signal is operational in resistant cells. Although some studies indicate that NF-κB can activate downstream of PI3K/Akt,(54,55) we do not know whether EGFR-PI3K/Akt pathway contributes to the activation of NF-κB in resistant cells.
Although our study is limited (only one cell line could be developed for this analysis), our recent observation from breast cancer tissues and genomic analysis of HER2+ breast cancer suggests that cancer cells undergo phenotype change during grade change (termed “progression through grade”) from HER2+ luminal B to HER2+ basal-like.(56,57) JIMT-1 (HER2+ basal-like) cells show constitutively activated NF-κB and nuclear accumulation of p65/RelA.(39) Thus, we surmise that by activating the NF-κB pathway and by progressive nuclear accumulation of p65/RelA, luminal B cells may be primed to obtain HER2+ basal-like phenotype. This might explain why both basal-like and HER2+ tumor cells show propensity for brain metastasis.(3,4) Our data lend to the “progression through grade” hypothesis.(57) We hypothesize that NF-κB activation may be a critical molecular event in the evolution of TZR in luminal B cells; potential pathways involved are illustrated in Figure 5. Our study also suggests that an early NF-κB activation and the nuclear accumulation of p65/RelA may be useful markers to identify metastasis-initiating cells.
In summary, we have shown that NF-κB is constitutively activated in HER2+ luminal B breast cancer cells that have acquired resistance to trastuzumab treatment without any chemotherapy or radiation treatment. Cells with an acquired resistance showed an increased sensitivity to the NF-κB inhibitor, and the combination of the NF-κB inhibitor plus trastuzumab has shown significantly greater inhibition of proliferation than either of the agents alone. Moreover, we showed that the NF-κB activation in resistant cells leads to an increase in cytokine and chemokine expression. We identified a potential mechanism of TZR where NF-κB activation through IKK kinase complex may be perpetuated. Our results further support NF-κB inhibition as a strategy for improving cell response to trastuzumab and investigating the level of NF-κB activation as a predictor of response to trastuzumab. Ongoing studies will examine the contribution of the IKK complex in the activation of NF-κB in human tissues from patients with advanced cancer who manifest brain metastasis. These studies are necessary to establish NF-κB and/or its regulators as new therapeutic targets in breast cancer brain metastasis.
Footnotes
Acknowledgments
We would like to thank Prof. Mark I. Greene, University of Pennsylvania, for providing T6–17 cells for the study. We would also like to thank Dr. Colleen Moody for her assistance in editing this article. This work was partly supported by NCI/NIH grants R01CA089481 and R01CA149425 (MIG/RM).
Author Disclosure Statement
The authors have no financial interests to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.