
Abstract
Premembrane (prM) is a viral protein of flavivirus, which is important for the generation of infectious virion and for virus infection to the host. However, the biological properties and function of the prM of Avian Tembusu virus (ATMUV) have scarcely been studied to date. Monoclonal antibodies (mAbs) are a powerful tool for functional analysis of viral protein. To produce a mAb against prM protein of ATMUV, the prM gene sequence was amplified by reverse transcription polymerase chain reaction (RT-PCR) and cloned into the prokaryotic expression vector pET-28a (+). The recombinant prM protein was successfully expressed in BL21 (DE3). Using the purified prM as immunogen in mice, three hybridoma cells secreting mAbs against prM protein were obtained. These mAbs showed a strong reaction with ATMUV-infected DF-1 cells and pEGFP-C3-prM transfected 293-T cells in both Western blotting analysis and immunofluorescence assay. The mAbs developed in this study will be useful tools for analysis of the prM protein functions on ATMUV infection and the interaction between prM and its host molecules.
Introduction
I
ATMUV-infected ducks mainly show a sudden drop in egg production of about 10%–90%, with other neurological symptoms, including paralysis and twisted neck. Hemorrhagic ovarian inflammation is the main pathological change in infected ducks.(4,5) ATMUV infects almost all species of duck. Other avian species such as goose, chicken, pigeon, and sparrow were also found to be infected.(6–8) In the past several years, the disease has caused great economic loss to the duck industries in China.
ATMUV belongs to the Ntaya virus group of flavivirus genus within the flaviviridae family.(1) The genome of ATMUV is a single-stranded positive-sense RNA with 10,990 nt in length, which possesses the typical genome structure as 5′-UTR-C-prM-E-NSI-NS2A-NS2B-NS3-NS4A-NS4B-NS5-UTR-3′. The whole flavivirus genome contains a big open reading frame of 10,278 nt encoding a single polyprotein, which is cleaved into three structural proteins (C, prM, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) by proteolytic enzymes.(9,10)
As a newly emerged disease, the functions of the ATMUV structural and nonstructural proteins are being identified. Currently, E protein of ATMUV has been primarily studied and was found to be able to induce neutralizing antibody in the host. Based on the E protein and its monoclonal antibodies (mAbs), a solid-phase competition enzyme-linked immunosorbent assay (ELISA) to detect antibodies against ATMUV has been established.(11) Nonstructural protein 1 (NS1) of flavivirus plays a role in virus maturation and mediates the host immune response to viral infection.(12,13) mAbs to NS1 of ATMUV were developed which will be useful for further functional analysis of NS1 of ATMUV.(14) However, few studies on the premembrane (prM) protein of ATMUV are reported. prM of flavivirus is a viral glycoprotein, interacting with E protein to facilitate the generation of infectious virion.(15) Antibody to prM is able to enhance dengue virus infection in the host.(16,17)
In this study, we successfully expressed prM protein of ATMUV in Escherichia coli. Using the purified recombinant prM as antigen, three mAbs against prM of ATMUV were obtained which will provide an effective tool for further study of prM protein function, the pathogenic mechanism of ATMUV, and control measures for ATMUV infection.
Materials and Methods
Virus strains and cells
The ATMUV YY1 strain was isolated from a duck farm in Zhejiang Province and maintained in our laboratory. DF-1 cells and human embryonic kidney 293-T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Gibco-BRL Life Technologies, Grand Island, NY). SP2/0 myeloma cell line was routinely maintained in our laboratory.
RNA extraction
The total viral RNA was extracted from YY1-infected DF-1 cells with TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions, and complementary DNA (cDNA) was synthesized using a PrimeScript first strand cDNA Synthesis Kit (TaKaRa Biotechnology Co., Ltd., Dalian, China).
Expression and purification of recombinant His tagged prM protein
A set of the following primers were designed to amplify the full length of ATMUV prM gene according to the genome sequence of YY1 strain (Genbank No. AB917088.1): prM-F, 5′-TA
The resulting plasmids were transformed into E. coli BL21 (DE3) cells and induced to express recombinant his-prM with 1 mM isopropyl-b-D-thiogalactopyranoside (IPTG). The cells were collected by centrifugation at 4000 × g for 5 minutes, then washed and resuspended in 1/10 the volume of phosphate-buffered saline (PBS), and lysed by sonication. After sonication, the lysates were centrifuged again to separate the supernatant and pellets, which were subject to SDS-PAGE to analyze the recombinant protein expression pattern. The protein expressed in the cell pellets were resuspended in buffer A provided in a Protein Purified Kit (QIAGEN GmbH; QIAGEN, Hilden, Germany), and the recombinant protein was purified using nickel-charged nitrilotriacetic acid (NTA)-agarose affinity resin. The protein concentration was determined with a Bicinchoninic Acid Assay Kit (Thermo, Waltham, MA). The purified protein was stored at −70°C for future use.
Preparation of monoclonal antibody against prM protein
Four six-week-old female BALB/c mice were immunized subcutaneously with an emulsion mixture containing an equal volume of complete Freund's adjuvant (Sigma-Aldrich, St. Louis, MO) and purified recombinant prM protein (200 μg prM per mouse). Two booster immunizations with the same dose of antigen emulsified with an equal volume of incomplete Freund's adjuvant (Sigma-Aldrich) were administered to the mice at 2-week intervals. Three days after the final immunization, the anti-prM antibody titer was determined by ELISA and the mouse with the highest titer was bled and sacrificed. The mouse splenocytes were collected and fused with SP2/0 myeloma cells. The hybridoma cells were cultured in RPMI-1640 medium containing HAT (hypoxanthine-aminopterin-thymidine) and HT (aminopterin-thymidine) for 10–14 days. The hybridomas were screened for antibody production against prM by ELISA. The hybridomas producing anti-prM antibodies were then subcloned four to five times by limiting dilution assay and confirmed by an indirect immunofluorescence assay (IFA) with ATMUV-infected DF-1 cells. The ascitic fluid containing the mAbs against prM was produced in paraffin-primed BALB/c mice. The isotype of the mAbs was determined using the SBA Clonotyping System-HRP system according to the manufacturer's protocol (Southern Biotechnology Associates, Birmingham, AL).
Enzyme-linked immunosorbent assay
Antibody titers of serum from prM immunized mice and monoclonal antibody titers in ascites and in supernatant of hybridoma cell culture were examined by ELISA. Purified recombinant prM protein was used as the coating antigens for the ELISA. One hundred microliters of the prM protein (2 μg/mL in 50 mM sodium carbonate buffer [pH 9.6]) was added into each well of a 96-well microtiter plate and then incubated overnight at 4°C. The plate was then washed thrice with PBST (0.25% Tween-20 in PBS) and incubated with mouse sera or mAbs for 1 hour at 37°C. After another washing, the plate was incubated with 100 μL of HRP-conjugated goat anti-mouse IgG (KPL, Gaithersburg, MD) (1:5000) for an additional hour at 37°C. The plate was then washed again, and the colorimetric reaction was developed using 3,3′,5,5′-tetramethylbenzidine for 10 minutes at 37°C. The reaction was stopped with 2 mol/L sulfuric acid, and the optical density was read at 450 nm.
In vitro transfection
To further analyze the reaction of the mAbs with ATMUV prM protein, the prM gene was constructed into eukaryotic expressing vector pEGFP-C3, and the constructed plasmid vector was named as pEGP-C3-prM. Twenty-four micrograms of the constructed plasmid with 60 μL of Lipofectamine 2000 (Invitrogen) were diluted in 1500 μL of DMEM without serum and antibiotics and then added into 293-T cell monolayers grown to 80% confluence in a 96-well cell culture plate. Twenty-four hours post-transfection, the cells were analyzed by Western blotting and IFA to determine the reaction of prM with the mAbs.
SDS-PAGE and Western blotting analysis
DF-1 cells infected with ATMUV or 293-T cells transfected with the pEGFP-C3-prM plasmid were collected and lysed in lysis buffer (1% SDS, 0.1% Triton X-100) on ice. Then the cell lysates or recombinant prM were mixed with sample loading buffer (P1016; Solarbio Science & Technology Co., Ltd., Beijing, China) and boiled for 5 minutes. The proteins were separated by standard SDS-PAGE gel and transferred onto nitrocellulose membranes (pore size 0.45 mm; GE Healthcare, Germany). The membranes were blocked in blocking buffer (PBS containing 5% skimmed milk) for 1 hour at room temperature and then rinsed with PBS thrice. The membranes were incubated with prM mAbs or positive serum to ATMUV infection in blocking buffer for 1 hour at room temperature. After being rinsed in PBS five times, the membranes were incubated with HRP-conjugated anti-mouse (KPL) or anti-duck IgG (KPL) secondary antibody for 1 hour at room temperature. The proteins were visualized with SungkyMaxi ECL Chemiluminescence Substrate (Sungky, Changzhou, China) under the conditions recommended by the manufacturer. Images were captured on a chemiluminescent imaging system (Cell Biosciences, Santa Clara, CA).
Indirect immunofluorescence assay
To detect the reactivity of the mAbs, DF-1 cells were infected with YY1 and 293-T cells were transfected with the pEGFP-C3-prM in a 96-well plate, respectively. The cells were then incubated at 37°C for 48 hours and then fixed with 4% paraformaldehyde. The fixed cells were incubated with mAbs or ATMUV-infected duck serum for 1 hour at 37°C. After being washed thrice with PBS, the cells were incubated with diluted anti-mouse Alexa Fluor 546 antibody (Life Technologies, Eugene, OR) or FITC-conjugated anti-mouse IgG secondary antibody (KPL) for 1 hour at 37°C. The stained cells were rinsed five times with PBS and then visualized using an LSM780 laser scanning confocal microscope (Zeiss, Oberkochen, Germany).
Results
Expression of recombinant prM protein in E. coli
To express the prM protein of ATMUV in E. coli, the 501 bp prM gene fragment was amplified from the cDNA of YY1-infected DF-1 cells and then constructed into a prokaryotic expression vector and transformed into BL21 cells for recombinant protein expression by induction with IPTG. As shown in the SDS-PAGE analysis, the protein was mostly expressed as inclusion bodies with the predicted molecular mass of around 20 kDa (Fig. 1A). In denatured conditions, the recombinant prM was effectively purified from the E. coli containing the pET-28a-prM plasmid (Fig. 1A). Western blotting analysis indicates that the purified prM could react with both mAb to His and serum from ATMUV-infected duck (Fig. 1B).
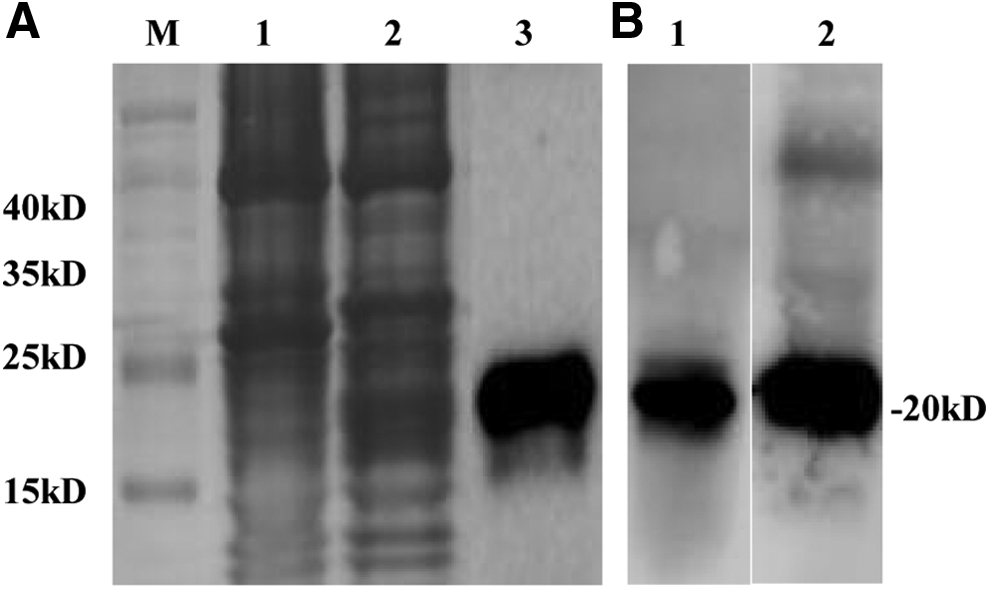
SDS-PAGE and Western blotting analysis of prM protein expressed in BL21 (DE3) Escherichia coli.
Production of the mAbs to prM
The purified prM protein was used as an immunogen to generate mAbs to prM protein. After three immunizations, antibody titers to prM protein in immunized mice reached 1:25,600 as determined by prM-ELISA. The spleen cells of the mouse with the highest antibody titer were fused with SP2/0 cells. After three subclonings, three hybridoma clone persistently secreted antibodies reacting with prM protein as indicated by both ELISA and IFA were obtained and named 1D5, 2G11, and 1G2, respectively. The ELISA titers of the supernatants of 1D5, 2G11, and 1G2 hybridoma cell culture and the ascites of three mAbs are summarized in Table 1. The subtypes of the three mAbs are all belonged to IgG2a, with Kappa light chains (Table 1).
ELISA, enzyme-linked immunosorbent assay; mAb, monoclonal antibody; prM, premembrane; ATMUV, Avian Tembusu virus.
Reactivity of the mAbs to prM
The reactivity of the three mAbs was tested against recombinant prM, ATMUV-infected DF-1 cell, or pEGFP-C3-prM transfected 293-T cells. As shown by Western blotting analysis, three mAbs all clearly reacted with purified recombinant prM or YY1-infected DF-1 cell lysates or transfected 293-T cell lysates showing the reaction bands with expected molecular mass of 20 kDa, 20 kDa, and about 45 kDa, respectively (Fig. 2). The three mAbs could also react with YY1-infected DF-1 cells and pEGFP-C3-prM transfected 293-T cells by IFA, while no immunofluorescence was observed in the infected DF-1 cells or pEGFP-C3-prM transfected cells when an unrelated mAb was applied (Fig. 3).

The reactivity of mAbs to prM analyzed by Western blotting. The recombinant His-prM, YY1-infected cell lysates (Infection), and pEGFP-C3-prM transfected cell lysates (Transfection) were subjected to Western blotting to react with mAbs 1D5, 2G11, and 1G2.
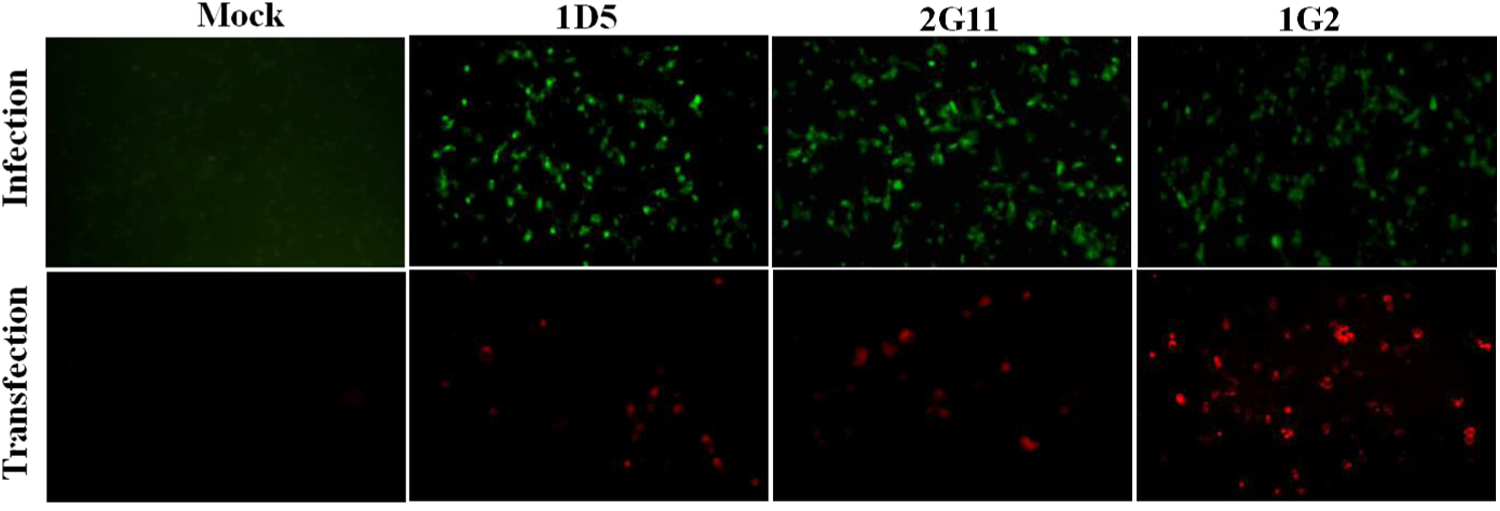
The reactivity of mAbs to prM analyzed by indirect immunofluorescence assay. YY1-infected DF-1 cells (Infection) and pEGFP-C3-prM transfected cells (Transfection) were incubated with mAbs 1D5, 2G11, and 1G2. YY1-infected cells and pEGFP-C3-prM transfected cells incubated with an unrelated mAb (mAb to His) served as control in Mock parallel experiments.
Discussion
Most of the viruses in flavivirus genus such as West Nile virus, dengue virus, tick-borne encephalitis virus, yellow fever virus, and Zika virus cause severe human diseases, which are transmitted by the bite from an infected mosquito or tick.(18,19) It has been reported that ATMUV was detected in mosquito and the sera from duck farmers contained antibody to ATMUV,(20,21) suggesting that as a member of flavivirus genus, ATMUV might have the potential to infect human beings. Furthermore, the molecular mechanism of ATMUV emerging as an avian infection agent in recent years is still not very clear. Understanding of the molecular basis is important to explain the evolution and pathogenic mechanism of ATMUV. So far, there is a lack of an in-depth study on the characterization and functional analysis of the ATMUV viral proteins.
In flavivirus, the membrane protein M is first synthesized as a precursor protein prM in the endoplasmic reticulum (ER), which is then associated with E protein to form the prM/E heterodimers, and presents on the surface of immature virion.(15) During transport of the immature virions through the ER and Golgi complex to the cell surface, the slightly acidic pH of the trans-Golgi network triggers dissociation of the prM/E heterodimers; the prM proteins were cleaved into the pr peptide and M protein by host furin. Upon dissociation of the pr peptide, mature virions are formed that are able to infect new cells.(15)
A previous study showed that mutations in the conserved region of prM led to the reduction of the infectious virions of tick-borne encephalitis virus in host cells, indicating the importance of prM protein during virus replication.(22) In contrast, the antibody to prM protein has been demonstrated to greatly enhance secondary flavivirus infection by binding to the Fc receptor of the host cells and leading to the immature virions getting infection capability.(17) Using the monoclonal antibody with virus replication-enhancing properties, the immunodominant antigenic site of dengue virus prM has been identified, which will be helpful for further understanding of the pathogenic mechanism of flavivirus.(23)
mAbs are valuable tools for characterization and functional analysis of viral proteins. In this study, we successfully expressed ATMUV prM recombinant protein using a prokaryotic expression system, which could be recognized by the ATMUV-infected duck serum (Fig. 1B). With the purified prM as immunogen, three mAbs 1D5, 2G11, and 1G2 have been obtained. These mAbs clearly react with ATMUV-infected cell lysates in Western blotting analysis (Fig. 2), indicating that the three mAbs recognize the linear B cell antigenic epitopes of prM protein.
In contrast, mAbs 1D5, 2G11, and 1G2 also react with ATMUV-infected cells and pEGFP-C3-prM transfected 293-T cells in IFA (Fig. 3), indicating that the mAbs produced in this study react with the native prM in the host cells. The IFA analysis showed that the prM protein reacted with the mAbs distributed as spots or particles in the infected or transfected cells, but not diffused in the whole cells. We speculate that the prM of ATMUV might be as described above for other flavivirus prM mainly localizing on certain organelles such as the ER and Golgi of host cells to have function on virus replication. With the mAbs against ATMUV prM, more studies should be carried out to further understand the function of prM.
In conclusion, three mAbs against the prM protein of ATMUV were developed and characterized in this study. These three mAbs may be an efficient tool to analyze the biological function of prM during ATMUV replication in the host cells and the interaction of prM with other viral protein such as E protein or host molecules. This is the first report on the production of a monoclonal antibody to ATMUV prM protein.
Footnotes
Acknowledgment
This work was supported by grant from the National Key Technology R&D Program of China (Grant No. 2015BAD12B01).
Author Disclosure Statement
No competing financial interests exist.