
Abstract
Podoplanin (PDPN), a type I transmembrane 36-kDa glycoprotein, is expressed not only in normal cells, such as renal epithelial cells (podocytes), lymphatic endothelial cells, and pulmonary type I alveolar cells, but also in cancer cells, including brain tumors and lung squamous cell carcinomas. Podoplanin activates platelet aggregation by binding to C-type lectin-like receptor-2 (CLEC-2) on platelets, and the podoplanin/CLEC-2 interaction facilitates blood/lymphatic vessel separation. We previously produced neutralizing anti-human podoplanin monoclonal antibody (mAb), clone NZ-1 (rat IgG2a, lambda), which neutralizes the podoplanin/CLEC-2 interaction and inhibits platelet aggregation and cancer metastasis. Human–rat chimeric antibody, NZ-8, was previously developed using variable regions of NZ-1 and human constant regions of heavy chain (IgG1) and light chain (kappa chain). Although NZ-8 showed high antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) against human podoplanin-expressing cancer cells, the binding affinity of NZ-8 was lower than that of NZ-1. Herein, we produced a novel human–rat chimeric antibody, NZ-12, the constant regions of which consist of IgG1 heavy chain and lambda light chain. Using flow cytometry, we demonstrated that the binding affinity of NZ-12 was much higher than that of NZ-8. Furthermore, ADCC and CDC activities of NZ-12 were significantly increased against glioblastoma cell lines (LN319 and D397) and lung cancer cell line (PC-10). These results suggested that NZ-12 could become a promising therapeutic antibody against podoplanin-expressing brain tumors and lung cancers.
Introduction
A
We previously produced a rat anti-human podoplanin monoclonal antibody (mAb), NZ-1.(5) NZ-1 possesses high specificity, high sensitivity, and high binding-affinity against podoplanin.(25,26) Because NZ-1 is highly internalized into glioma cell lines, and is also well accumulated into tumors in vivo, NZ-1 is a suitable candidate for therapy against malignant gliomas.(27) By its neutralizing activity, NZ-1 also inhibited the tumor cell-induced platelet aggregation and cancer metastasis.(12) We constructed a single-chain antibody variable region fragment using NZ-1 (NZ-1-scFv).(28) NZ-1-scFv was then fused to Pseudomonas exotoxin A (NZ-1-scFv-PE38). NZ-1-scFv-PE38 exhibited significant activity against glioblastoma and medulloblastoma cells, and demonstrated tumor growth delay in D2159MG and D283MED in vivo tumor models.
Furthermore, we produced a human–rat chimeric anti-podoplanin antibody (NZ-8) from NZ-1.(29) NZ-8 possesses high antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Furthermore, NZ-8 inhibits the growth of podoplanin-expressing tumors in vivo, and also suppresses hematogenous metastasis of podoplanin-expressing tumors. In contrast, the binding affinity of NZ-8 is lower than that of NZ-1.
Herein, we produced a novel human–rat chimeric antibody, NZ-12, the constant regions of which consist of IgG1 heavy chain and lambda light chain.
Materials and Methods
Cell lines
CHO-K1 was obtained from the American Type Culture Collection (ATCC, Manassas, VA). LN319 and D397 were provided by Dr. Kazuhiko Mishima (Saitama Medical University, Saitama, Japan) and Dr. Darell D. Bigner (Duke University Medical Center, Durham, NC), respectively. PC-10 cells were purchased from Immuno-Biological Laboratories Co., Ltd. (Gunma, Japan). CHO-K1 and PC-10 were cultured in RPMI 1640 medium (Nacalai Tesque, Inc., Kyoto, Japan), and LN319 and D397 were cultured in Dulbecco's modified Eagle's medium (DMEM) (Nacalai Tesque, Inc.), supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.) at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Antibodies
A rat anti-human podoplanin mAb (NZ-1) and a human–rat chimeric anti-human podoplanin antibody (NZ-8) were developed as described previously.(5,27,29,30) For the generation of human–rat chimera anti-human podoplanin (NZ-12), the appropriate VH of a rat NZ-1 antibody and CH of human IgG1 were subcloned into the pCAG-Neo (Wako Pure Chemical Industries Ltd., Osaka, Japan), and VL of a rat NZ-1 antibody and CL of human lambda chain were subcloned into pCAG-Ble vectors (Wako Pure Chemical Industries Ltd.). Antibody expression vectors were transfected into CHO-K1 cells using Lipofectamin LTX (Thermo Fisher Scientific, Inc.). Stable transfectants of CHO/NZ-12 were selected by cultivating the transfectants in medium containing 1 mg/mL G418 (Wako Pure Chemical Industries Ltd.) and 0.5 mg/mL Zeocin (InvivoGen, San Diego, CA). CHO/NZ-12 cells were cultivated in CHO-S-SFM II medium (Thermo Fisher Scientific, Inc.). NZ-12 was purified using Protein G-Sepharose (GE healthcare Bio-Sciences, Pittsburgh, PA). Human IgG was purchased from Sigma-Aldrich Corp. (St. Louis, MO).
Determination of the binding affinity by flow cytometry
LN319 cells (2 × 105 cells) were resuspended with 100 μL of serially diluted antibodies (NZ-1, NZ-8, or NZ-12; 0.02–100 μg/mL) followed by FITC-labeled anti-human IgG or Oregon green-labeled anti-rat IgG (Thermo Fisher Scientific, Inc.). Fluorescence data were collected using a cell analyzer (EC800; Sony Corp.). The dissociation constants (KD) were obtained by fitting the binding isotherms using the built-in one-site binding models in Prism software.
Preparation of effector cells
The preparation methods of effector cells have been described previously.(31) Human peripheral blood mononuculear cells (MNCs) were obtained from leukocytes, which were separated from peripheral blood of healthy donors. The human study was approved by the Ethics Committee of Tokushima University.
Antibody-dependent cellular cytotoxicity
ADCC was determined with 51Cr release assay.(31,32) Target cells were incubated with 0.1 μCi of 51Cr-sodium chromate at 37°C for 1 hour. After washing with CRPMI1640 (RPMI1640 medium including 10% FBS) three times, 51Cr-labeled target cells were placed in 96-well plates in triplicate. Effector cells and antibodies were added to the plates. After 6 hours incubation, 51Cr release of the supernatant from each well (100 μL) was measured using a gamma counter (PerkinElmer, Waltham, MA). The percentage of cytotoxicity was calculated using the following formula: % Specific lysis =(E−S)/(M−S) × 100, where E is the release in the test sample, S is the spontaneous release, and M is the maximum release.
Complement-dependent cytotoxicity
CDC was evaluated by 51Cr release assay as described previously.(31,32) Target cells were incubated with 51Cr-sodium chromate (0.1 μCi) for 1 hour at 37°C. After that, the cells were washed with CRPMI1640. 51Cr-labeled cells were incubated with baby rabbit complement (Cedarlane, Ontario, Canada) at a dilution of 1:4 and antibodies (1 μg/mL) for 6 hours in 96-well plates. After incubation, the supernatant including 51Cr was measured using a gamma counter. The percentage of cytotoxicity was calculated as described above.
Statistical analyses
The statistical significance of differences in in vitro data was analyzed by standard Student's t-test. In this study, p-values less than 0.05 were considered significant in all experiments.
Results and Discussion
Development of a novel human–rat chimeric anti-podoplanin antibody (NZ-12)
The sensitivity of NZ-8 is lower than that of NZ-1.(29) NZ-8 was generated by fusing the VH of rat antibody (NZ-1) with CH of human IgG1 and fusing VL regions of NZ-1 with CL of human kappa because almost all chimeric antibodies and humanized antibodies are comprised of kappa light chains.(33,34) In contrast, NZ-1 possesses lambda light chain, the rare isotype found among rat IgGs (<5%).(35) Therefore, we subsequently generated the human–rat chimeric antibody (NZ-12) by fusing the VH of rat antibody (NZ-1) with CH of human IgG1 and fusing VL regions of NZ-1 with CL of human lambda.
NZ-12 demonstrated high sensitivity against podoplanin-expressing cancer cell lines in flow cytometry, and its reactivity was shown to be much higher than that of NZ-8 (data not shown). KD of NZ-1 was determined to be 1.2 × 10−9 M by flow cytometry (Fig. 1). Using the same methods, KD of NZ-8 was determined to be 7.3 × 10−7 M (Fig. 1), indicating that the binding affinity of NZ-8 was diminished following conversion from rat mAb to a human–rat chimeric antibody. In contrast, KD of NZ-12 was determined to be 8.5 × 10−9 M by flow cytometry (Fig. 1), indicating that the binding affinity against a podoplanin-expressing cell line (LN319) became much higher following conversion from a human kappa light chain (NZ-8) to a human lambda light chain (NZ-12).
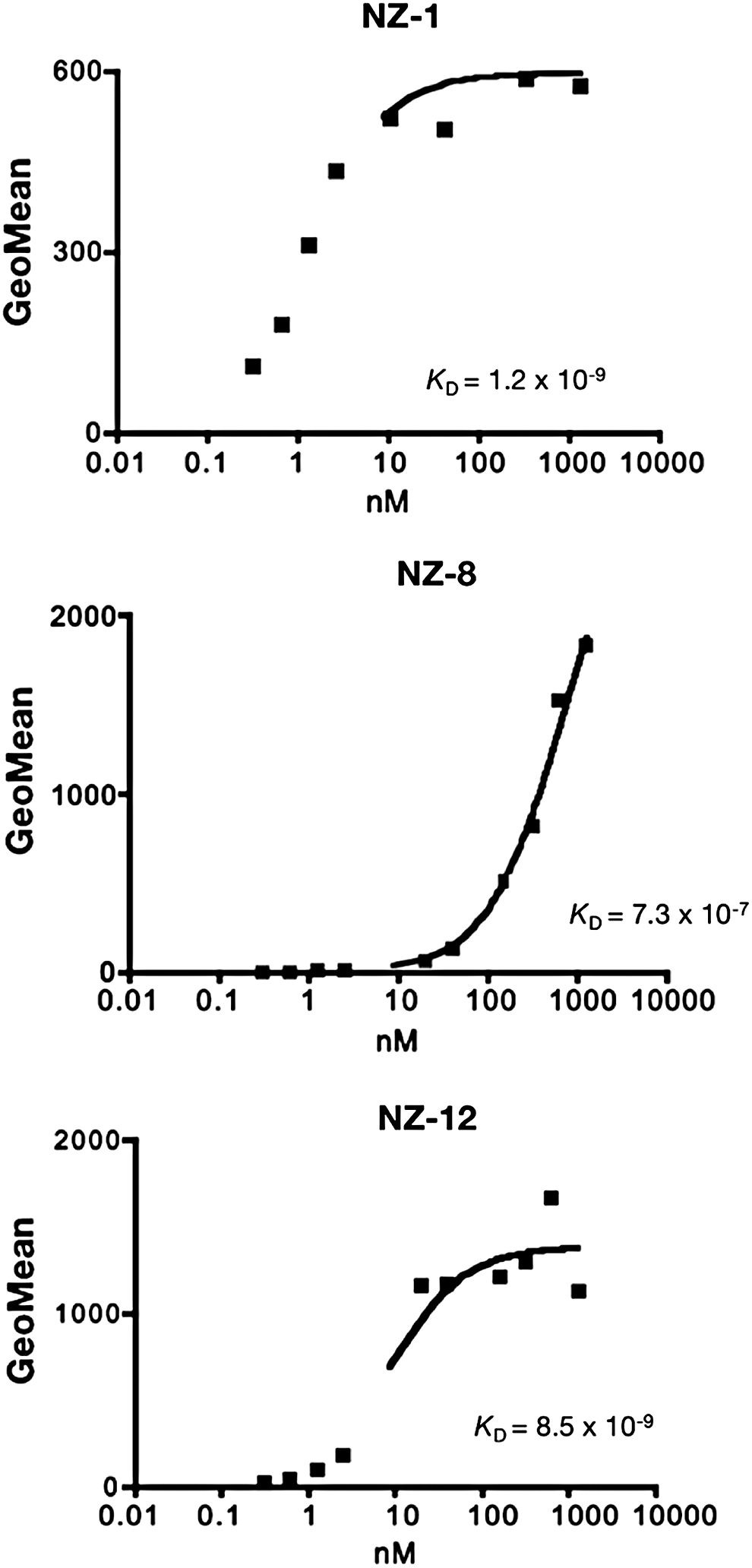
Determination of binding-affinity using flow cytometry. LN319 (2 × 105 cells) was resuspended at 100 μL of serially diluted NZ-1, NZ-8, or NZ-12 (0.02–100 μg/mL).
ADCC and CDC mediated by anti-podoplanin antibodies
To apply targeted therapy to podoplanin, we further assessed whether anti-podoplanin antibodies could induce ADCC against podoplanin-expressing cell lines mediated by human MNC as effector cells. We compared the ADCC and CDC activities against NZ-1, NZ-8, and NZ-12 using LN319, D397, and PC-10 cell lines. ADCC against podoplanin-expressing cell lines was not exhibited by NZ-1 using human MNC, whereas NZ-8 and NZ-12 showed the induction of significant level of ADCC mediated by human MNCs against LN319 and D397 glioblastoma cells and PC-10 lung cancer cells (Fig. 2A). Furthermore, ADCC by NZ-12 was higher compared with NZ-8 particularly against LN319 and D397 glioblastoma cells. Interestingly, CDC was induced by NZ-12 against all podoplanin-expressing cell lines, and this was significantly higher than that of NZ-8 (Fig. 2B).
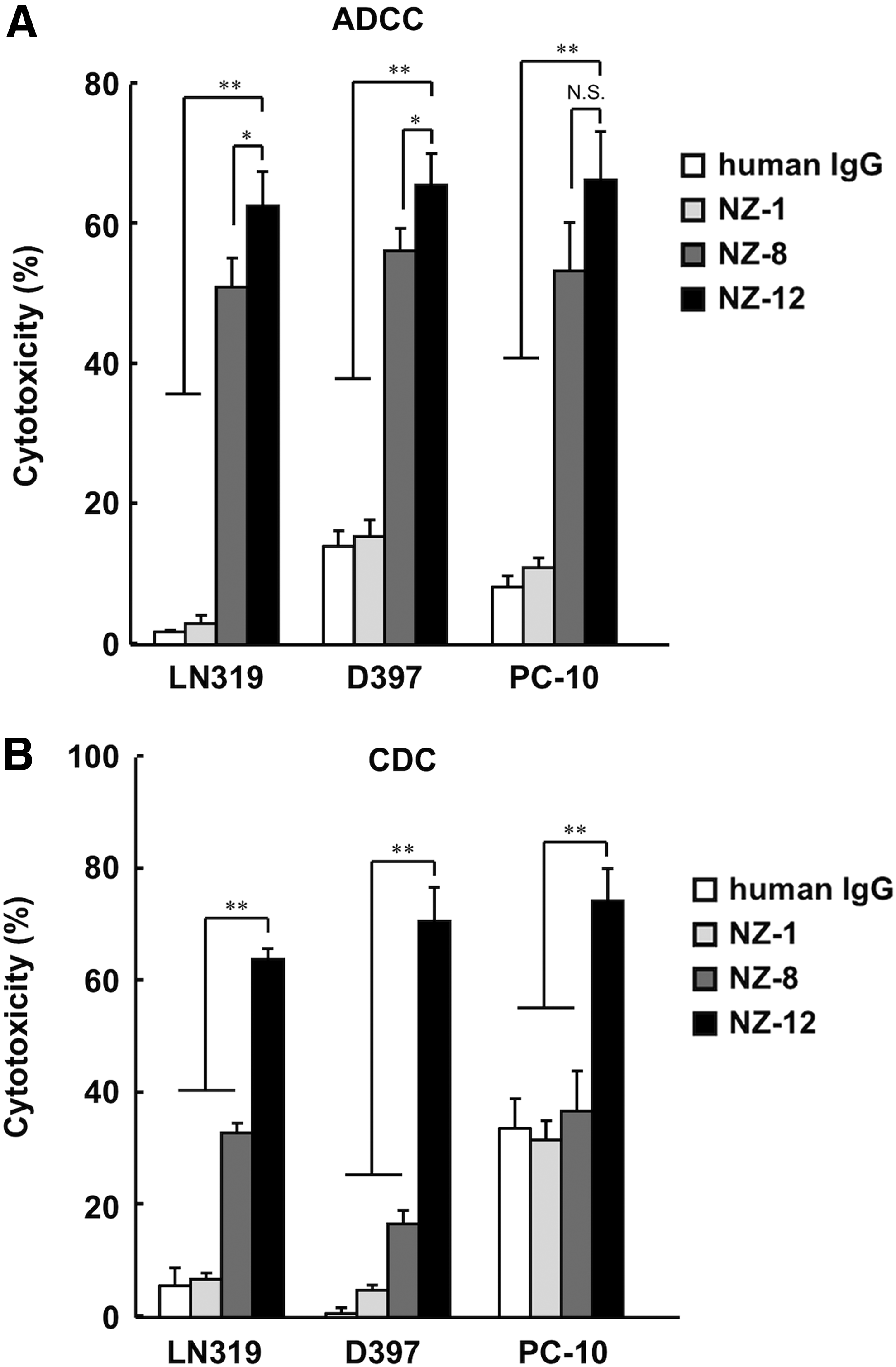
ADCC and CDC activities of anti-podoplanin antibodies.
Taken together, NZ-12 could be useful for antibody therapy against human podoplanin-expressing cancers. In the future, NZ-1/NZ-12 could be further applied to the novel anti-tumor reagents, including T cells and viruses.(36,37)
Footnotes
Acknowledgments
We thank Miyuki Yanaka, Takuro Nakamura, Noriko Saidoh, and Kanae Yoshida for their excellent technical assistance. This work was supported in part by the Basic Science and Platform Technology Program for Innovative Biological Medicine from Japan Agency for Medical Research and development, AMED (Y.K.), by the Platform for Drug Discovery, Informatics, and Structural Life Science (PDIS) from AMED (Y.K., T.M.), by the Project for utilizing glycans in the development of innovative drug discovery technologies from AMED (Y.K.), by the Translational Research Network Program from AMED (Y.K.), by the Regional Innovation Strategy Support Program from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (Y.K.), by JSPS KAKENHI Grant Number 26440019 (M.K.K.) and 16K10748 (Y.K.). This work was performed in part under the Cooperative Research Program of Institute for Protein Research, Osaka University, CR-16-05 and by the Grant for Joint Research Project of the Institute of Medical Science, the University of Tokyo. The authors would like to thank Enago (
Author Disclosure Statement
No competing financial interests exist.