
Abstract
Hepatitis C virus (HCV) is a significant health threat that has been extensively investigated worldwide. Improving the sensitivity and specificity of laboratory tests for screening and early diagnosis of HCV in a relevant population is an effective measure to control the spread of HCV. To build a more reliable diagnostic method for HCV, we expressed gene fragments of HCV-NS3 linked to a carrier, pET28a, and then transformed this vector into Escherichia. coli. The produced recombinant NS3 protein with a molecular weight of 38 kDa, which was purified through Ni-chelating affinity chromatography, was used to immunize BALB/C mice, which generated a serum antibody titer of 1:160,000 against the immunogen. Three positive monoclonal isolates (2A5, 2A6, and 5B12) were screened and established. Western blot and enzyme-linked immunosorbent assay (ELISA) results of these monoclonal cells show that each could specifically recognize the recombinant protein. Antibodies 2A5 and 2A6 were developed into an ELISA sandwich antibody pair for the recombinant protein. The detection sensitivity of our developed ELISA was 1.6 ng/mL, with a linear range of 2.5–80 ng/mL (R2 = 0.998). Serum NS3 ELISA results show that the average value in the healthy group, liver disease group, and hepatitis C group was 3.71, 7.28, and 13.11 ng/mL, respectively. The positive rates of HCV-NS3 protein in the liver disease group and hepatitis C group was 17.2% and 41.7%, respectively. Detection of HCV-NS3 antigen can be used as an auxiliary test for anti-HCV antibody detection, thus reducing leakage detection and providing a reliable basis for clinical practice.
Introduction
H
HCV is a single-stranded RNA virus ∼9.6 kb in length. Its genome includes an open reading frame (ORF) that is located on the central gene. More than 3000 amino acids in the ORF encode a single precursor polyprotein, which is cleaved by both host and viral proteases into 10 HCV proteins, each with individual functions. These 10 proteins, core protein, envelope protein 1, envelope protein 2, p7, and nonstructural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B, were named based on their different functions.(4)
Currently, there are two primary methods for the diagnosis and management of HCV infection. The first is the detection of anti-HCV-specific antibodies present in patient serum samples. This approach is fairly rapid, but is not definitive and cannot be used during the early phases of acute infection (<6 months). The second detection method is a molecular assay that detects viral nucleic acids.(5) This method is much more sensitive and provides a definite result. Nevertheless, anti-HCV antibody detection is the recommended first-line testing. Generations of anti-HCV detection tools have been developed, but they all have significant shortcomings, including high false positive rates and a lack of sensitivity to detect the antibodies in the window period, limiting its use in acutely infected patients. In addition, detection of anti-HCV antibodies cannot distinguish whether the patient has recovered or whether the patient shows an active infection. Thus, anti-HCV detection methods cannot be used to monitor patient treatment.(2,6) Given these limitations, an HCV nucleic acid test (NAT) is necessary to confirm active HCV infection and guide disease management.(7) Multiple NATs for both qualitative and quantitative HCV RNA detection are available. The International Clinical Practice Guidelines recommend the use of real-time polymerase chain reaction (PCR) for HCV RNA detection and quantification, and it has been widely used in clinical virology laboratories.(8) An HCV-RNA positive result indicates that viral replication is taking place in vivo, whereas an HCV-RNA negative result demonstrates that the virus had been cleared. Therefore, PCR should be used in addition to anti-HCV testing for further diagnosis of anti-HCV positive samples. Unfortunately, HCV detection by PCR is time consuming, labor intensive, and costly, making its use prohibitive in the developing world.(9,10)
NS3, 1 of the 10 HCV proteins, spans 1027–1659aa of the >3000aa polyprotein encoded by HCV and has a molecular weight of ∼69 kDa.(11) NS3 has a variety of biochemical functions, such as serine protease activity near the 1/3 N-terminal (1–180aa) and nucleoside triphosphate and helicase activity in nearly two-thirds of the C-terminus.(12–14) It is also of critical importance for viral replication, and has become an important target for new research in the diagnosis, treatment, and prevention of HCV.(11,15) NS3 protein appears early in HCV infection and maintains sustained levels for as long as the virus remains active. In addition, its strong affinity with a specific antibody suggests a greater advantage in laboratory detection of HCV. Xie et al. showed perfect specificity and a high sensitivity of HCV-NS3 antigen in human serum using an enzyme-linked immunosorbent assay (ELISA).(16) Another study by Sillanpää et al. demonstrated with Western blot analysis that the NS3 protein was recognized in 68% of HCV patient serum samples.(17)
Our aim is to provide a better, more reliable diagnostic tool for HCV. To achieve this aim, we developed a population of monoclonal antibodies with high sensitivity to HCV-NS3 and then developed a highly sensitive ELISA for diagnostic testing. The development of the NS3 protein, and subsequent monoclonal antibodies and an ELISA platform will allow for additional tools to research and facilitate the diagnosis of HCV in at-risk or infected patients.
Materials and Methods
Materials
The pET28a vector and E. coli BL21 were acquired from Takara (Dalian, China). Primers were synthesized at Genewiz (Suzhou, China). BCA Protein Assay Kit, Dulbecco's modified Eagle medium (DMEM), and fetal bovine serum were purchased from Thermo Fisher. The Rapid Mouse Isotyping Kit-Gold Series was purchased from RayBiotech (Guangzhou, China). HiTrap Protein A HP was purchased from GE Healthcare (Atlanta, GA). Goat Anti-Mouse IgG-HRP was purchased from Medi biological (Liaoning, China). All other chemicals and reagents used throughout the experiments were from Sigma Aldrich (St. Louis, MO).
Production and purification of recombinant HCV-NS3 protein
Amplification of the HCV-NS3 gene (Gly100aa-Ile303aa) [Uniprot Accession # F4YQS5] encoding the HCV-NS3 protein was carried out using the oligonucleotide primers NS3-F (5′-AAACATATGGGCAGCTCGGACCTTTAC-3′) and NS3-R (5′-AAACTCGAGTCAGATGCCCAAAATGGTAGT-3′), which amplifies a 612 bp region encoding the NS3 epitope sequence. The recombinant gene was linked to the expression vector (pET28a), and was then expressed in E. coli. The bacteria were collected by centrifugation and broken by ultrasonic wave. The protein was then purified with a His-Bind Nickel column. The purified material was then separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Coomassie Blue to assess its molecular mass. The concentration of purified protein was determined with a BCA assay kit (Sigma, St. Louis, MO).
Preparation and characterization of antibodies
BALB/c mice were purchased from Southern Medical University (Guangzhou, China). Mice were then injected subcutaneously with the antigen and TiterMax adjuvant (Atlanta, GA) mixture, and then reimmunized every 2 weeks for a total of four immunizations. The immunization was deemed successful when the mouse antiserum titer reached 1:80,000, at which time they were prepared for development of hybridoma cell lines.
SP2/0 myeloma cells were cultured in DMEM supplemented with 10% fetal bovine serum before fusion. Three days after the booster immunization, splenocytes were isolated from immunized mice and fused with SP2/0 cells. Mouse splenocytes were mixed with SP2/0 cells at a ratio of 5:1, and then 1 mL 50% polyethylene glycol (Sigma) was slowly added for 1 minute. After letting the mixture stand for 60 seconds, 3 mL of DMEM was added for 3 minutes, and then 25 mL of DMEM was added into the mixture for the next 3 minutes. Next, the fused cells were centrifuged and resuspended in hypoxanthine [H], aminopterin [A], thymidine [T] (HAT) selection medium containing 20% serum. The cell suspension was transferred into 96-well culture plates coated with feeder cells from the peritoneal cavity of nonimmunized mice and then grown in a specified incubator (Thermo) with 5% CO2 at 37°C. Three days after fusion, half of the medium was replaced with fresh HAT medium, and 1 week later the entire supernatant was switched to HT medium containing 20% serum. The culture supernatants were then tested for activity by indirect ELISA after a week.
Positive hybridoma cells were subcloned two to three times by limiting dilution. Three hybridoma cell lines were then established and prepared in mouse ascites, followed by purification by affinity chromatography on a protein-G column. In our study, indirect ELISA was used to test antiserum titer, the positive hybridoma, and the final monoclonal antibody titer. The subtypes of antibodies were identified by the Rapid Mouse Isotyping Kit-Gold Series according to the manufacturer (Raybiotech).
Indirect ELISA
Microtiter plates were coated overnight with 1 μg/mL of recombinant HCV-NS3 in 10 mM of sodium carbonate buffer (pH 9.6) at 4°C, washed three times in phosphate buffered saline (PBS) containing 0.05% Tween-20 buffer (PBST), and then blocked with 5% skim milk in PBS at 37°C for 2 hours. After a final wash, 100 μL of antiserum, culture supernatant, or antibody diluted in PBS were added to each well and incubated for 1 hour at 37°C. Then, 100 μL of a secondary antibody (HRP-goat antimouse antibody) at 1:10,000 diluted in PBS was added to each well and incubated for 1 hour. The plates were washed six times, and then the tetramethylbenzidine (TMB) substrate was added for 5 minutes at room temperature in the dark. The TMB reaction was then quenched with 2 mol/L sulfuric acid (H2SO4), and the optical density (OD) was read at 450 nm with a microplate reader (Biotek).
Specificity test of monoclonal antibody by Western blotting
Recombinant HCV-NS3 protein was separated on SDS-PAGE, and then transferred to a nitrocellulose membrane (Millipore Corp., Bedford, MA). Subsequently, the membrane was blocked with 5% skim milk in PBST for 2 hours at 37°C. The membrane was washed four times with PBST and incubated with the primary antibodies at room temperature for 1 hour. After washing, the membrane was incubated with IRDye 800CW donkey antimouse IgG (H+L) (Odyssey) for visualization with the Odyssey infrared imaging system (LI-COR).
Development of HCV-NS3 sandwich ELISA
Monoclonal antibodies were labeled with NHS-Biotin (Raybiotech, Atlanta, GA) for development of a sandwich ELISA. Fifty micrograms of purified antibody was dialyzed overnight at 4°C with precooled 1× PBS. Antibodies were collected and 10 μg NHS-Biotin was added, then mixed for 4 hours at 37°C. The mixture was dialyzed again to remove any free biotin. Biotinylated antibodies were collected and the protein concentration was determined with a BCA assay kit.
A sandwich ELISA platform was developed as follows. Polystyrene microtiter plates (Beaver, Suzhou, China) were coated with 100 μL anti-HCV-NS3 monoclonal antibody (1 μg/mL) overnight at 4°C and blocked with 2% BSA for 2 hours at 37°C. After three washes with PBST, a standard solution (at 100, 50, 25, 12.5, 6.25, 3.125, 1.5625, and 0 ng/mL concentrations) was added to the wells. Wells were washed as before, and then incubated with 100 μL biotinylated antibody (1 μg/mL) at 37°C for 1 hour. The plate was washed again, and HRP–streptavidin was added into each well and incubated for 1 hour at 37°C. Finally, 100 μL TMB substrate was added to each well and incubated at 37°C for 10 minutes, after which the reaction was quenched with 50 μL of H2SO4 (2 mol/L), and then the absorbance was measured at 450nm (BIO-RAD). We used recombinant HCV-NS3 protein to generate a standard curve to measure binding potential. The detection limit of this method was defined as the concentration of HCV-NS3 consistent with the absorbance of the blank value plus 3 standard deviations above the blank signal. Zero concentration was repeated 20 times, then the average value (M) and standard deviation (SD) were calculated. The detection sensitivity was determined by M + 2SD.
Sample detection
All sera of people, aged from 15 to 72 years old with the average age of 37.6 ± 2.8 years, were collected by The Third Affiliated Hospital of Sun Yat-sen University in the period from September 2015 to October 2016. Twelve cases of hepatitis C group were positive for HCV antibody. Twenty-nine cases of liver disease group were diagnosed for liver disease such as cirrhosis, fatty liver, or liver fibrosis, but were negative for HCV antibody. Healthy serum was donated by 10 healthy people. Serum samples were measured by Sandwich ELISA. The content of HCV-NS3 in the serum sample was calculated according to the standard curve.
Results
Recombinant HCV-NS3 protein
We analyzed HCV-NS3 sequences using DNASTAR software and identified many potentially immunogenic epitopes (Fig. 1). A hydrophilicity >0, a surface probability >1, and flexibility are important indicators for predicting the epitope. With this information, we selected a region of the protein, glycine 100—isoleucine 303 (Uniprot Accession # F4YQS5), for expression and development of our monoclonal antibodies.
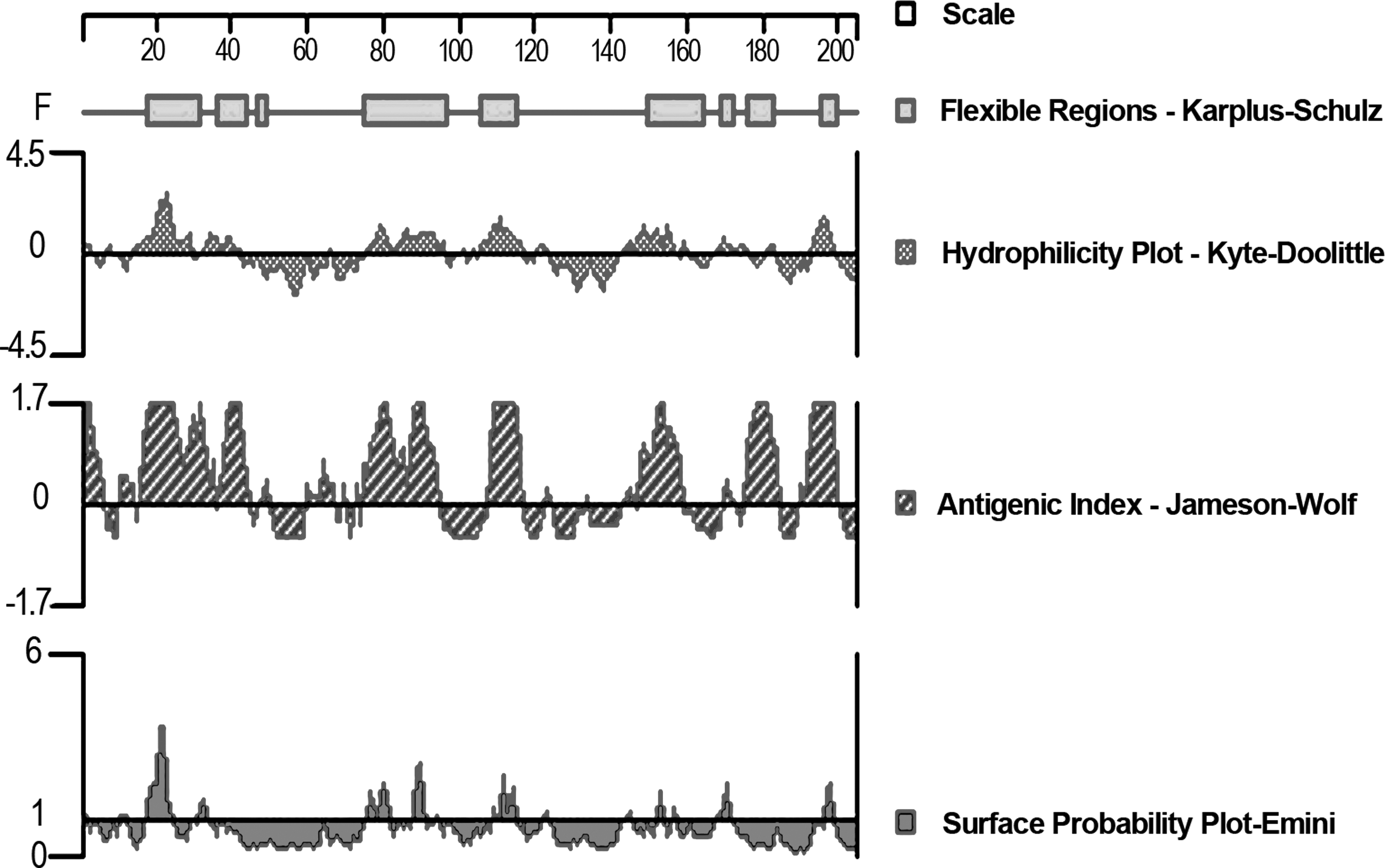
Analysis of the characteristics of the NS3 sequence. Hydrophilicity, surface probability, flexibility, and antigenic index of HCV-NS3 predicted by Dnastar software. HCV, hepatitis C virus.
A recombinant expression plasmid, pET28a (+)-HCV-NS3, was transformed into E. coli to prepare recombinant HCV-NS3 protein. The protein was primarily expressed in insoluble inclusion bodies. As such, after inclusion body lysis, the recombinant proteins were purified by his-tag immobilized metal ion affinity chromatography. As expected, the target protein band in the eluted fraction was identified at 38 kDa, as demonstrated with SDS-PAGE analysis (Fig. 2A). The purified protein was further analyzed with a Western blot using an anti-his antibody, which identified a clear and distinct band at 38 kDa (Fig. 2B).

Purification of HCV-NS3 recombinant protein.
Preparation of anti-HCV-NS3 antibodies
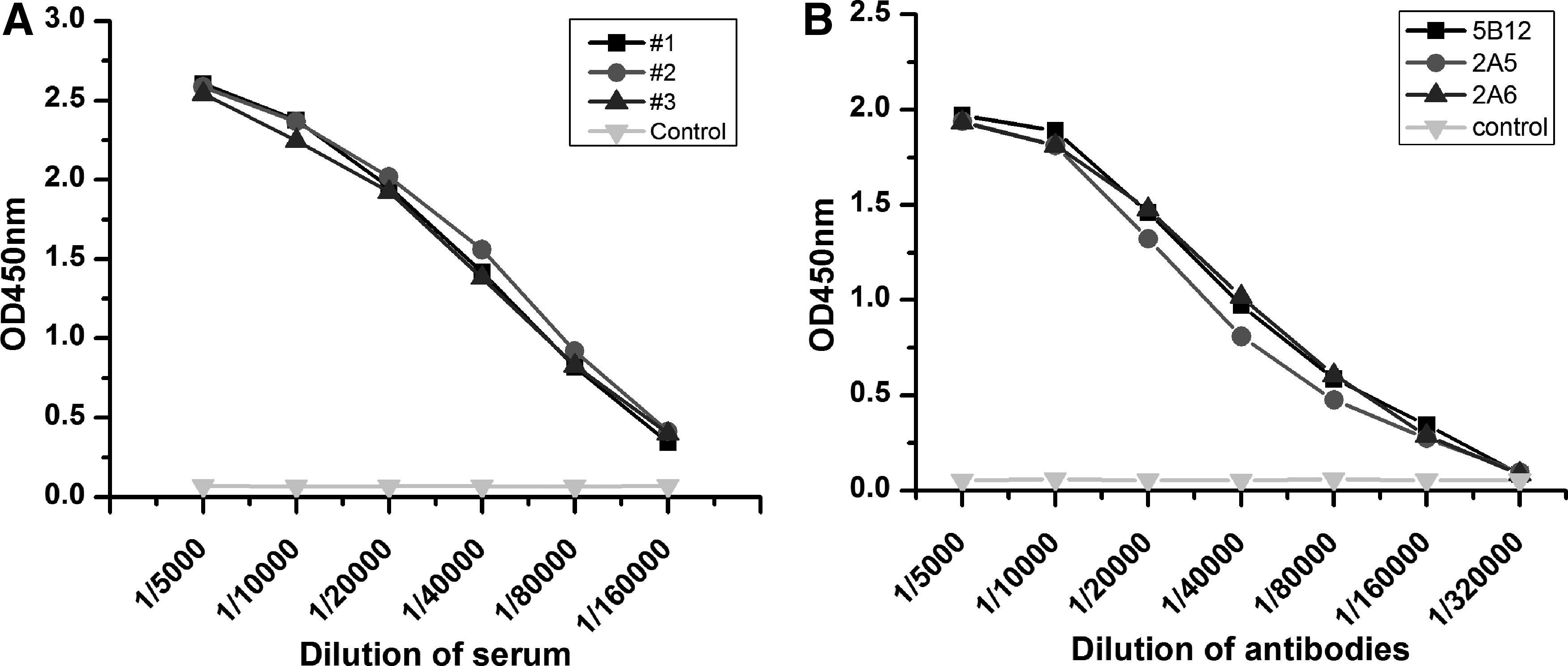
The purified protein was then used as the immunogen to develop antibodies through subcutaneous injection. Mice were immunized biweekly either three or four times until the antiserum titer was sufficient. As seen in Figure 3A, titers of three immunized mice achieved 1:160,000 (OD values at 1:160,000 exceed the twofold OD value of the control). In this study, mouse #1 was chosen for splenocyte extraction and fusion for monoclonal antibody development. After fusion and subsequent screening, we obtained three monoclonal cell lines that stably secrete antibody against HCV-NS3 (clones 2A5, 2A6, and 5B12).
All antibodies developed were prepared from ascites in mice. Ten-week-old BALB/c mice were injected intraperitoneally with pristane 1 week before injection of 0.5 mL of hybridoma culture suspensions (1 × 106 hybridoma cells). Seven days after hybridoma injection, ascites were collected and purified by saturated ammonium sulfate precipitation and Protein G affinity column, successively.
Characterization of antibodies
Indirect ELISAs were performed to determine antibody titers in the immunized mice. Microtiter plates were coated with NS3 protein (100 ng/well). They were then probed with antiserum (diluted 2× starting at 1/5000), or with monoclonal antibodies starting at 1 mg/mL and diluted 1/5000, 1/10,000, 1/20,000, 1/40,000, 1/80,000, 1/160,000, and 1/320,000. HRP-goat antimouse IgG was used as a secondary antibody and diluted 1/10,000. The titer of the purified monoclonal antibodies reached nearly 1/320,000, as shown in Figure 3B. To determine the isotype of the monoclonal cells, Rapid Mouse Isotyping Kit-Gold Series strips were used (Raybiotech). The analysis indicated that all three antibodies belong to the IgG1 subtype. Western blot analysis revealed a single, specific band at ∼38 kDa corresponding to the size of the HCV-NS3 immunogen (Fig. 4).
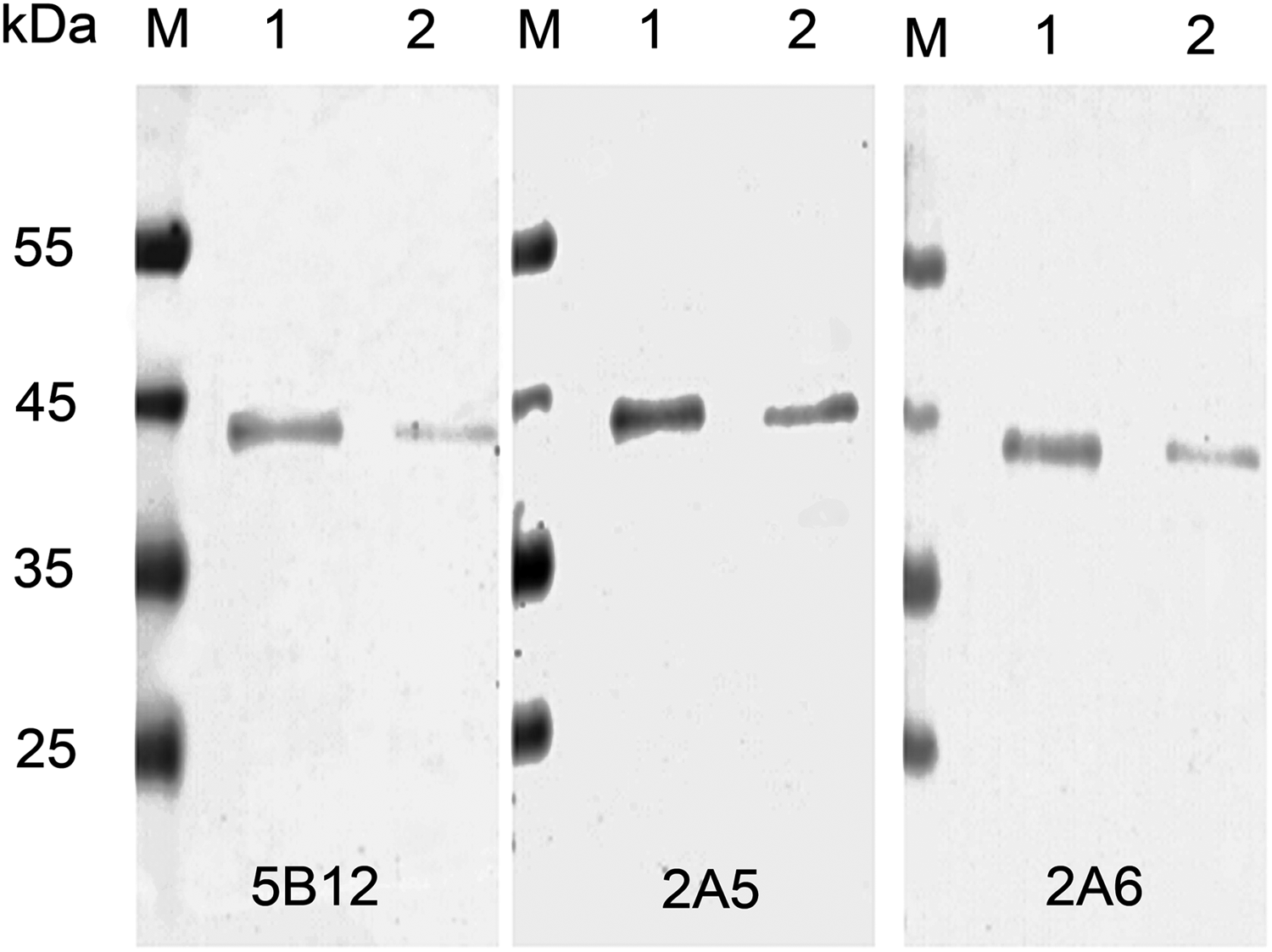
Immunological reaction of monoclonal antibodies and HCV-NS3 protein was analyzed by Western blot. Equal amounts of HCV-NS3 protein were loaded. Lane 1, 20 ng protein; lane 2, 10 ng protein.
Cross-reactivity analysis
To determine the specificity of the antibodies to their target protein, we screened the antibodies against multiple related and distant proteins. The three monoclonal antibodies were tested against recombinant HCV-NS3, HCV-core, and HIV-P24 (RayBiotech). As previously mentioned, different proteins were coated on the plates, and examined by indirect ELISA. As shown in Figure 5, all three monoclonal antibodies were found to only recognize the target protein HCV-NS3.
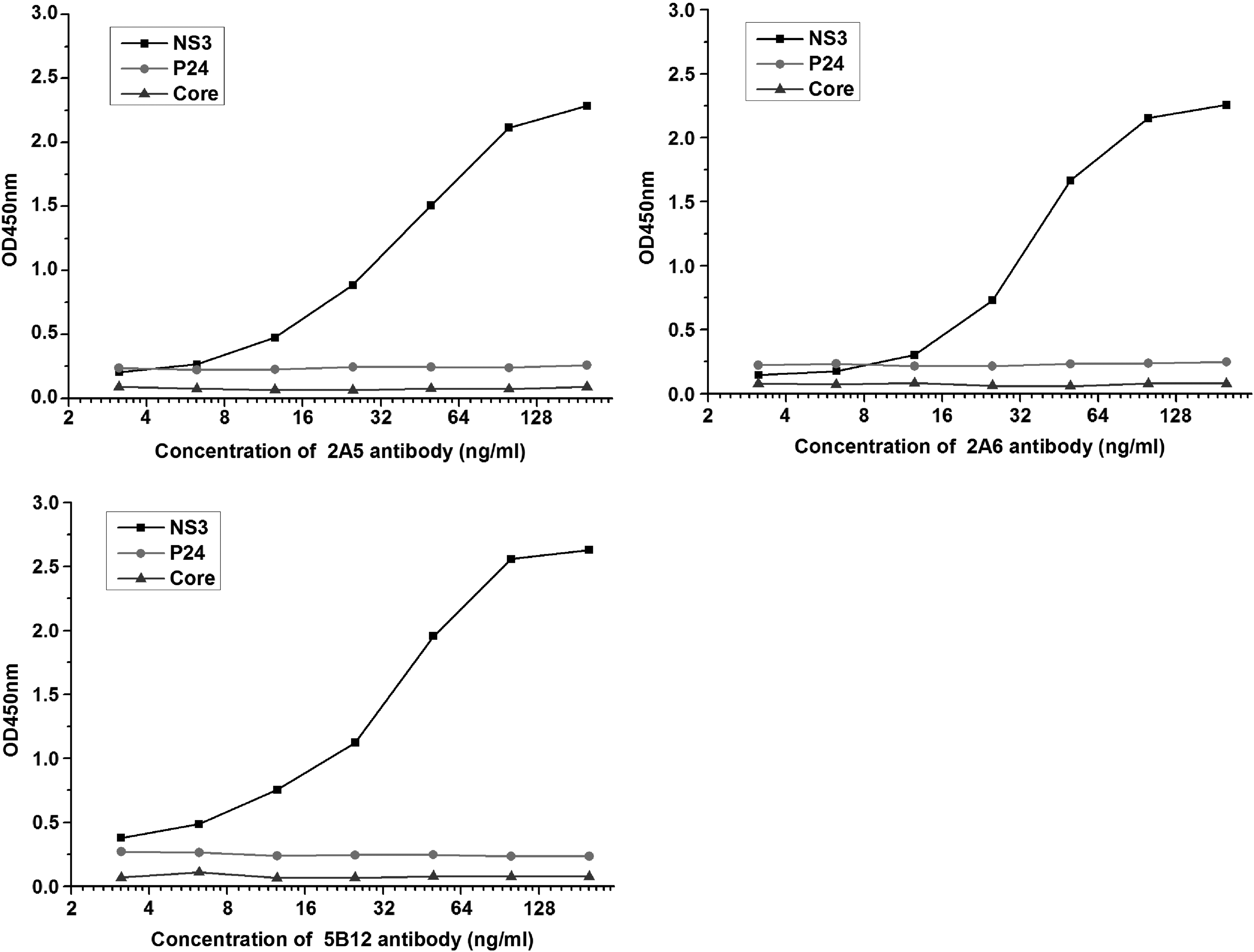
Specificity analysis of antibodies by indirect ELISA. The graph shows the level of specific binding of the antibodies to the coated proteins.
Establishment of sandwich ELISA
Using the three developed monoclonal antibodies, we next set out to see whether they could be paired together in a sandwich ELISA setting. Various combinations of the monoclonal antibodies were used as capture or detection antibodies. As shown in Table 1, the most obvious difference is in the detection value of 1.5625 and 0 ng/mL (M + 2SD). In addition, the detection value of this combination has a good linear trend (R 2 = 0.99). We identified the pairing of 2A6 and 2A5 as capture and detection pairs, respectively, as having excellent sensitivity. To finalize the development of a pair and an ELISA testing platform, we adjusted our protocol to further optimize sensitivity. By switching our blocking reagent to 2% bovine serum albumin with 1.5% trehalose and increasing the incubation time of HRP–streptomycin to 45 minutes, we were able to substantially improve the platform. Three parallel experiments were carried out for testing purposes and to develop the average values and standard deviation for the kit (Fig. 6A). This testing found an excellent standard curve, with a strong linear relationship from 2.5 to 80 ng/mL, with an R 2 = 0.998 (Fig. 6B), and a detection limit of 1.6 ng/mL.
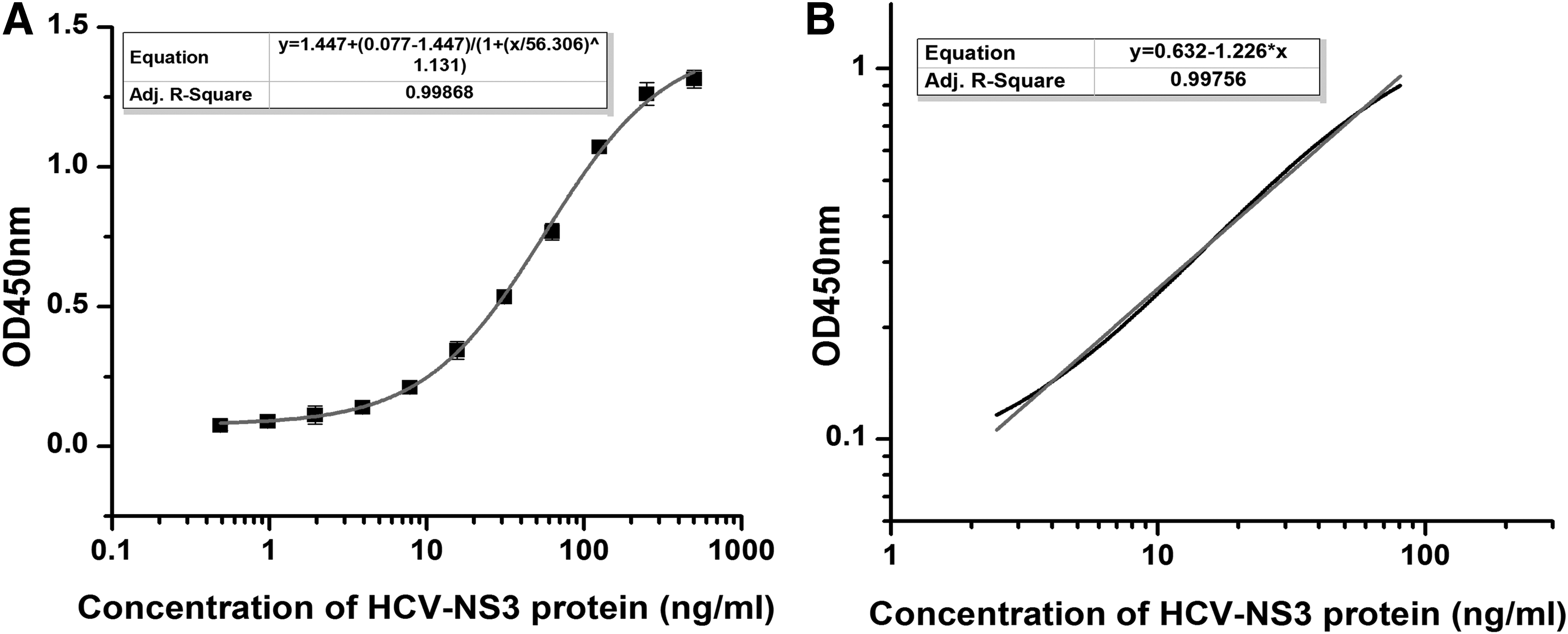
Development of HCV-NS3 sandwich ELISA with the 2A5 and 2A6 antibody pair.
Cab, capture antibody; Dab, detection antibody; HCV, hepatitis C virus; M, the average value of 20 repetitions of 0 ng/mL; R, the correlation coefficients of linear regression equations; SD, the standard deviation of 20 repetitions of 0 ng/mL.
Analysis of NS3 protein in human serum samples
Human serum samples (healthy (n = 10), hepatitis C (n = 12), and liver disease (n = 29) samples) were tested, and the serum NS3 concentration was calculated based on the standard curve of the recombinant NS3 protein. The results show that the average value in the healthy group, liver disease group, and hepatitis C groups was 3.71, 7.28, and 13.11 ng/mL, respectively (Fig. 7). These data were analyzed with a nonparametric test and a p-value <0.05 was considered significant. The expression of NS3 in the hepatitis C group compared with that in the healthy group was significantly different (p < 0.01), but there was no significant difference in expression between the hepatitis C and liver disease groups. In the hepatitis C group, some cases show a low level of HCV-NS3 expression, suggesting that the results of the HCV-NS3 protein and the antibody detection have some difference, Southeast Asian Journal of Tropical Medicine and Public Health. Here, the average level of serum HCV-NS3 of healthy people was considered negative, whereas the detection value was more than twice the negative value and is considered positive. Five out of 12 cases of hepatitis C were positive. The positive rate was 41.7%. The positive rate of HCV-NS3 in the 29 liver disease patients was 17.2%. These five cases of antibody negative but antigen positive were subsequently tested by PCR and the results all showed positive.
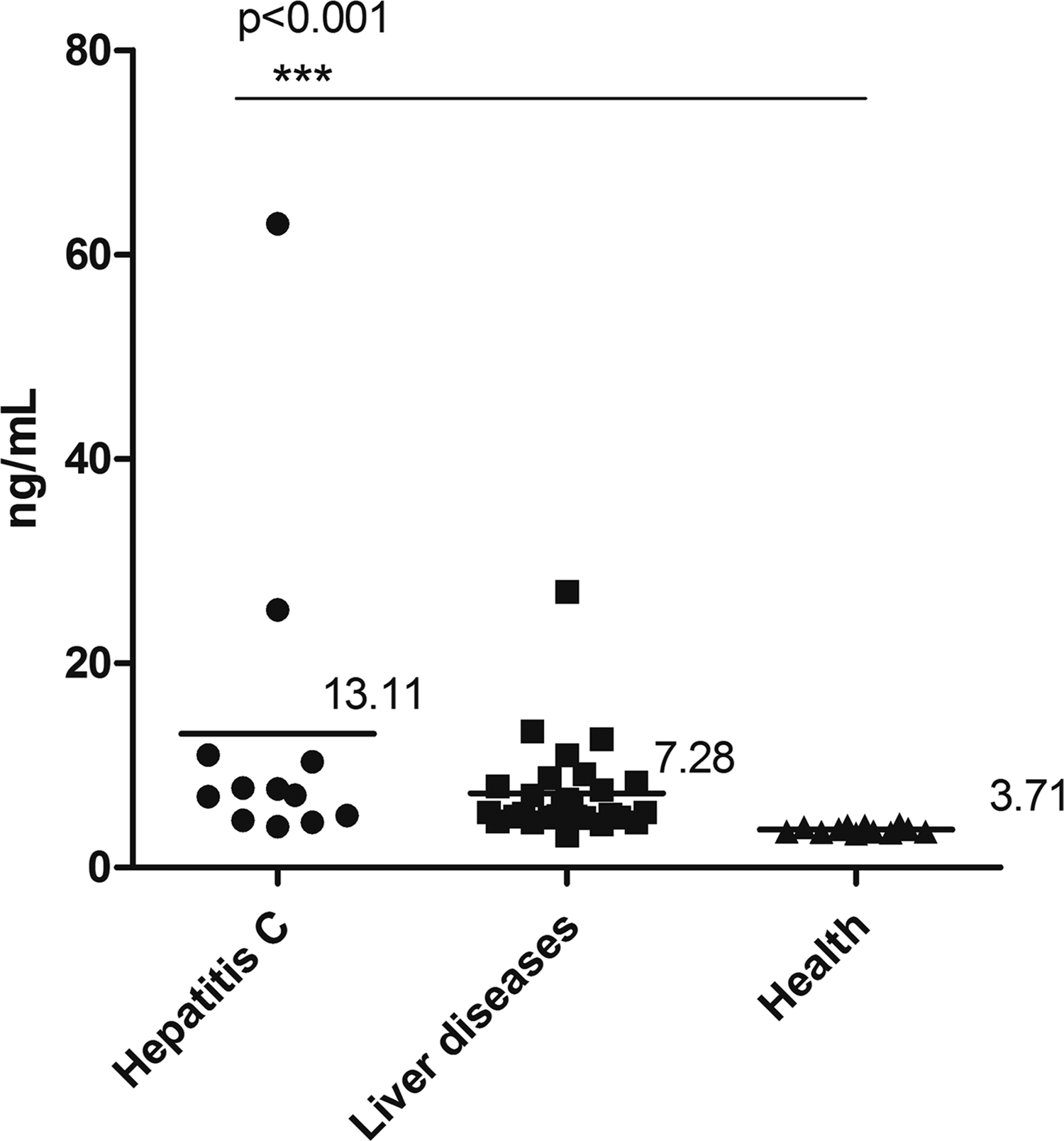
Analysis of serum NS3 levels with our developed sandwich ELISA. Average NS3 level in hepatitis C group is 13.11 ng/mL (n = 12), average NS3 level in liver disease group is 7.28 ng/mL (n = 29), and average NS3 level in health group is 3.71 ng/mL (n = 10).
Conclusion
With >170 million people infected with HCV globally, and no vaccine to protect against hepatitis C viral spread, a platform for the diagnosis and treatment of hepatitis C is essential.(18,19) This is especially true for a virus like HCV that causes chronic hepatitis, liver cirrhosis, hepatocellular carcinoma, and other liver diseases.
NS3 plays an important role in the viral infection life cycle, as it contains viral proteins serving as a serine protease and an RNA helicase. These two features have recently become important targets for antiviral therapy, as they are both necessary factors for replication and translation of the HCV genome.(20,21) NS3 is crucial to anti-HCV diagnostic reagents since it is both critical for the life of the virus and for generating a protein that is highly immunogenic. This finding of antigenicity is supported by recent studies that show that NS3 can generate a humoral immune response, and may therefore be of use in both diagnostics and therapies.(15,22,23)
In this study, we describe the generation of a recombinant HCV-NS3 protein and its use in monoclonal antibody development. The monoclonal antibodies produced were then used to find a compatible ELISA pair. Through screening, we found one pair of monoclonal antibodies that recognizes different epitopes, and that together had a sensitivity of 1.6 ng/mL and a linear range of detection between 2.5 and 80 ng/mL (R 2 = 0.998). Serum NS3 protein expression in the hepatitis C group was significantly increased compared with expression in the healthy group (p < 0.01), but was not significantly different compared with expression in the liver disease group as 5 out of 29 showed HCV-NS3 positive in the liver disease group. However, these five sera were subsequently tested by PCR and the results all showed positive. It can be seen from the results that there are some limitations in the clinical application of HCV protein alone, but it can be used as an auxiliary test for anti-HCV antibody detection, thus reducing leakage detection and providing a reliable basis for clinical practice. In the future, we plan to do additional clinical validation with our antibody pair with a larger sample size. In conclusion, we have developed and produced the raw materials needed for immunoassay development (such as ELISA, colloidal gold immunochromatographic strip assay, or chemiluminescence immunoassay), which could be crucial for future development of diagnostics and therapeutics for HCV detection.
Footnotes
Acknowledgments
We would like to express our thanks for the support to RayBiotech innovative research fund, UK-China (Guangzhou) Healthtech Open Innovation (2014Q-P037), Guangdong Provincial Science and Technology SME Technology Innovation Fund Program (2015A010101492), The Industrial Technical Project of Guangzhou Collaborative Innovation Major Projects (201604010116), The Science and Technology Project for People's Livelihood of Guangzhou Collaborative Innovation Major Projects (201604020159), Guangzhou Health Care Collaborative Innovation Major Projects (201604020012), and Guangzhou Pearl River Nova Program (201610010083).
Author Disclosure Statement
No competing financial interests exist.