
Abstract
Cadherin-17 (CDH17) is highly expressed in gastric cancer and is thus considered to be a good target for antibody therapy. CDH17 is classified as a nonclassical cadherin, in that it is composed of seven extracellular cadherin domains. We generated anti-CDH17 monoclonal antibodies (mAbs) which recognize the extracellular domain of CDH17. Competitive assay using AGS, a gastric cancer cell line, cells revealed that five selected anti-CDH17 mAbs recognize different epitopes on CDH17. As AGS cells were shown to exhibit broad expression pattern of CDH17 by flow cytometry, we separated three clones with a low (10,000/cell), medium (50,000/cell), and high (200,000/cell) expression level, designating them as AGSlow, AGSmed, and AGShigh, respectively. The mAbs, coupled with saporin, exhibited effective cytotoxicity to AGShigh, but poor cytotoxicity to AGSlow. By contrast, the immunotoxin cocktail using the three clones D2101, D2005, and D2008, which recognize different epitopes, exhibited efficient cytotoxicity, even to the AGSlow group. The effect of the immunotoxin cocktail is synergistic, as the combination index was demonstrated to be below 1.0, as calculated by the method of Chou and Talalay using CalcuSyn software. These results suggest that the immunotoxin cocktail targeted to multiple epitopes has synergistic effects on low expression level cells, which expand the applicable range of immunotoxin therapy for cancer.
Introduction
T
In previous studies, knockdown of CDH17 inhibited the growth of human gastric cancer cell by inactivation of Wnt/β-catenin(12) or Ras/Raf/MEK/ERK(13) signaling. The administration of an antibody against CDH17 suppressed hepatocellular carcinoma tumor xenograft in mice by inactivating the Wnt/β-catenin pathway.(14,15) Based on these accounts, CDH17 has potential not only as a diagnosis marker but also as a therapeutic target.
Gastric cancer is associated with a poor prognosis and is the third leading cause of cancer-specific death worldwide.(16) The positive rate of CDH17 expression with immunohistochemical staining is reportedly 74% in gastric cancerous tissues.(17) Thus, it is valuable for gastric cancer treatment for the purpose of developing an antibody drug conjugate (ADC) or immunotoxin therapy targeting CDH17.
ADCs are promising cancer therapeutics, as two clinically successful ADCs, ADCETRIS (brentuximab vedotin) and KADCYLA (ado-trastuzumab emtansine), are in clinical use. There are over 50 different ADCs in clinical development.(18) The effort to improve ADCs is in vigorous progress. ADC efficacy depends on target antigen expression, target internalization, antibody affinity, cytotoxic payload, linker stability, and conjugation type.(19) The most commonly utilized payload is tubulin-targeting antimitotic agents such as the maytansinoids, which are 1000-fold more powerful than standard drugs and thus are characterized by potentially serious side effects, resulting in a relatively narrow therapeutic window.(18) In the effort to improve ADC efficacy, cocktails of immunotoxins have been shown to exhibit a higher cytotoxic activity(20–22) by targeting several different antigens.
In this study, we applied a monoclonal antibody (mAb) cocktail for one target protein with multiple epitopes. We generated anti-CDH17 mAbs recognizing different epitopes on CDH17. Three of those mAbs were conjugated indirectly with saporin, a ribosome-inactivating protein, to obtain immunotoxins. Using established mAbs, we separated three types of the commercially available gastric cancer cell line AGS, that is, subclones (AGSlow, AGSmed, and AGShigh), which exhibited different CDH17 expression levels. Each immunotoxin exhibited sufficient cytotoxic activity against AGShigh expressing a high level of CDH17 (200,000/cell), although the cytotoxic activity was poor against AGSlow with a low CDH17 expression level (10,000/cell).
We demonstrated that the cocktail of different epitope-recognizing immunotoxins has synergistic cytotoxic effects against AGS cells. Thus, the multidomain targeting immunotoxin cocktail exerts a positive effect against the low level CDH17 expressing cells. This method should serve as an effective therapy for mixed tumors with different expression levels of target proteins.
Materials and Methods
Reagents
RPMI-1640, anti-FLAG M2 antibody, and ProClin300 were purchased from Sigma-Aldrich Japan (Tokyo, Japan). Protein G–Sepharose was purchased from GE Healthcare Japan (Tokyo, Japan). Ultralow IgG fetal bovine serum (FBS) and EZ-Link™ Sulfo-NHS-LC-Biotin were purchased from Thermo Fisher Scientific K.K. (Kanagawa, Japan). Block ACE was purchased from Dainippon Sumitomo Pharma (Osaka, Japan). Goat anti-mouse IgG (H+L) antibody, horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Fc fragment specific) antibody, FITC-conjugated anti-mouse IgG (Fc fragment specific), and Alexa Fluor 647-conjugated anti-mouse IgG (H+L) antibody were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). TMB (3,3′,5,5′-tetramethylbenzidine) Soluble Reagent and TMB Stop Buffer were purchased from ScyTek Laboratories (Logan, UT). PMMA beads were obtained from Sapidyne Instruments, Inc. (Boise, ID). Magnosphere™ MX200/carboxyl beads were obtained from JSR Life Sciences Corporation (Ibaraki, Japan). Streptavidin-saporin (Streptavidin-ZAP) was purchased from Advanced Targeting Systems (San Diego, CA). The Cell Counting Kit (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). Anti-His-tag mAbs (OGHIS) was purchased from Medical & Biological Laboratories Co., Ltd. (Aichi, Japan). The Human Cadherin-17 Antibody (MAB1032) was purchased from R&D systems (Minneapolis, MN). QIFIKIT was purchased from Dako (Carpinteria, CA). cOmplete® protease inhibitor cocktail was purchased from Roche diagnostics (Tokyo, Japan). Control IgG (B8109B, an anti-gp64 mAb) was established in our laboratory.
Cell lines
AGS cells were obtained from the American Type Culture Collection (ATCC), while MKN74 cells were obtained from the Japanese Collection of Research Bioresources (JCRB). The cell lines were cultured in RPMI 1640 medium containing 10% FBS, penicillin, and streptomycin at 37°C with 5% CO2. Single AGS cell cloning was performed by limiting dilution method, and three types of AGS cell lines with different CDH17 expression levels were designated as AGSlow, AGSmed, and AGShigh.
Preparation of human Cadherin-17 expressing budded baculovirus
The CDH17-expressing budded baculovirus (CDH17-BV) was generated as previously described.(23,24) Briefly, the gene of N-terminal FLAG and C-terminal Myc tagged CDH17 (NM_001144663) was optimized for codon usage for Sf9 and cloned into pUC57 vector by GenScript. The gene was cut with the restriction enzymes XbaI and KpnI and then inserted into the pFastBac 1 vector. Using the Bac-to-Bac baculovirus expression system, the recombinant baculovirus was generated according to the manufacturer's instructions. The recombinant baculovirus was collected from Sf9 culture media by centrifugation at 40,000 g for 40 minutes and resuspended with phosphate-buffered saline (PBS).
Anti-human Cadherin-17 mAbs
The recombinant baculovirus expressing human CDH17 (CDH17-BV) was immunized directly into gp64 transgenic mice and then the isolated spleen cells were fused with myeloma cells, as described.(23,24) Anti-CDH17 mAb producing hybridomas were selected by their reactivity to CDH17-expressing cancer cells, as determined by enzyme linked immunosorbent assay (ELISA) and flow cytometry. To obtain mAbs in culture supernatant, these hybridomas were cultured with 10% ultralow IgG FBS containing RPMI-1640 medium. After 2 weeks of culture, anti-CDH17 mAbs were purified from the culture supernatant using Protein G-Sepharose affinity chromatography. The purity of the mAbs was confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis, and the concentration of the mAbs was determined by absorbance at 280 nm.
Preparation of biotinylated antibodies and immunotoxin
mAbs were biotinylated with EZ-Link® Sulfo-NHS-LC-Biotin (Thermo Scientific) in accordance with the manufacturer's instructions, and the biotinylated mAbs were separated with a PD-10 desalting column (GE healthcare). For immunotoxin preparation, streptavidin-saporin was mixed with biotinylated mAbs in equal molar concentrations for 30 minutes at room temperature.
Immunoblotting
AGS cells were washed with PBS and dissociated with ethylenediaminetetraacetate (EDTA) in PBS. The pelleted cells were resuspended with 10 mM Tris-HCl, pH 8, containing protease inhibitor cocktail (cOmplete; Roche), and broken by passing a 26-gauge needle through them. The broken cells were centrifuged for 5 minutes at 660 g and then the supernatant was centrifuged for 30 minutes at 20,000 g. The pellet was resolved on SDS-PAGE and blotted onto a nitrocellulose membrane. Nonspecific binding was blocked using Block ACE and then the blots were probed with primary antibodies in reaction buffer (40% Block ACE and 0.05% ProClin300 in TBS). After washing with TBS-T (10 mM Tris-buffered saline containing 0.05% Tween 20), the blots were incubated with a HRP-conjugated goat anti-mouse antibody (Fc fragment-specific) diluted in reaction buffer and visualized using SuperSignal West Dura Extended Duration Substrate.
Flow cytometry and CDH17 quantification
Cells were dissociated with 2 mM EDTA in PBS, and 105 cells/well were plated on a round bottomed plate. After centrifugation of the plate at 2000 rpm for 2 minutes, the supernatants were removed, and the primary antibodies diluted with the dilution buffer (1% BSA and 0.1 mM EDTA in PBS) were added. After 1-hour incubation at 4°C, the cells were washed with dilution buffer and reacted with FITC-conjugated anti-mouse IgG diluted with dilution buffer for 1 hour at 4°C. Finally, cells were washed with dilution buffer and analyzed by flow cytometry (Guava®easyCyte™ Plus System; Millipore). The cell surface expression of CDH17 was quantified on the AGS cell lines using QIFIKIT according to the manufacturer's instruction.
Cell ELISA
Cells were dissociated with 2 mM EDTA in PBS and resuspended in culture medium. The cells were plated on poly
For the measurement of antibody binding activity, the plates were incubated with a primary antibody at various concentrations in reaction buffer for 1 hour. After washing thrice with 0.05% Tween 20 in saline, the plates were incubated for 1 hour with a HRP-conjugated anti-mouse IgG (Fc fragment-specific) antibody.
For the competition assay, the plates were incubated with a proper concentration of biotinylated antibody and competitive antibody in reaction buffer for 1 hour. After washing thrice, the plates were incubated with HRP-conjugated streptavidin.
After washing five times with 0.05% Tween 20 in saline, the enzymatic reaction was visualized with TMB Soluble Reagent. After the reaction was stopped with TMB Stop Buffer, the absorbance was measured at 450 nm using a microplate reader (Biotrak II; GE Healthcare, Piscataway, NJ).
Epitope mapping of anti-CDH17 mAbs
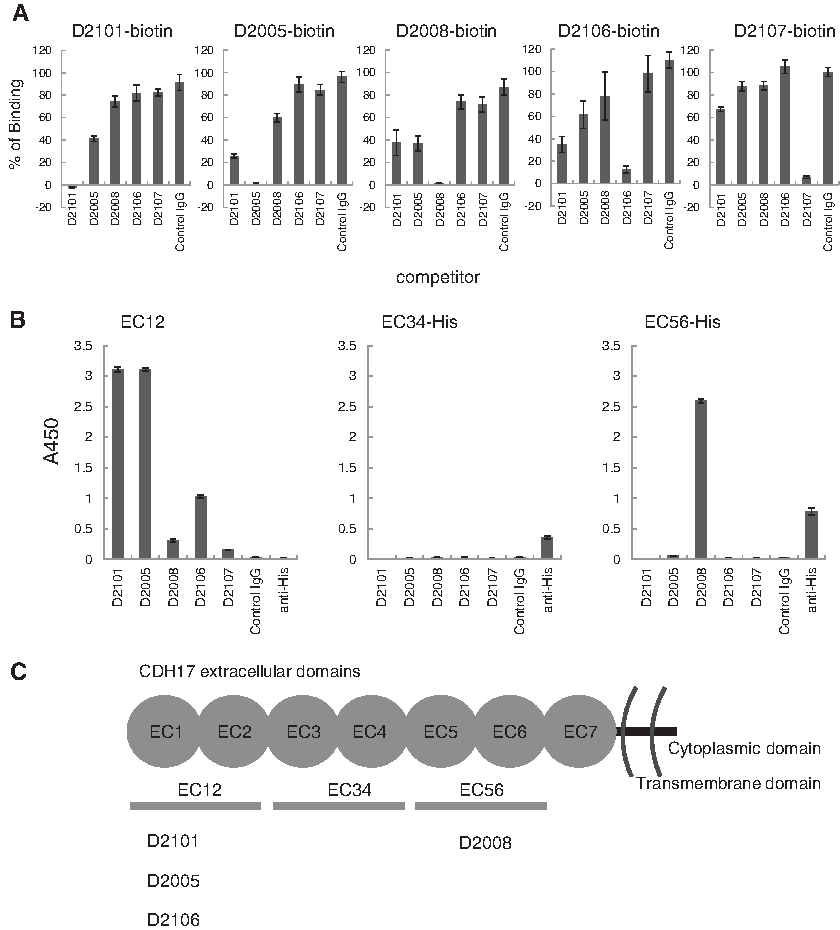
To determine the binding sites of the anti-CDH17 mAbs, two consecutive extracellular domains (from EC12, EC34, and EC56, as shown in Fig. 3C) containing recombinant fragments of CDH17 were generated in an Escherichia coli expression system. The fragments EC12, EC34, and EC56 are composed of the amino acid residues 23–236, 245–449, and 450–667 of human CDH17, respectively. Both the EC34 and EC56 constructs contained an additional Met residue at the N-terminus and a Leu-Glu-His6 sequence at the C-terminus. The expression vector for EC12 was constructed with an N-terminal His6-tag following a SUMO-tag for purification and efficient expression. The sequence of EC12 was cloned into a Champion pET SUMO vector using an In-Fusion® HD Cloning Kit (Clontech) with the forward primer 5′-CAGGAGGGAAAGTTCTCTGGTCC-3′ and reverse primer 5′-CTCGGTCACGATGATGTCCAC-3′. The expression vectors for EC34 and EC56 were constructed with a C-terminal His6-tag for purification. The sequences of EC34 and EC56 were cloned into a pET28b vector between the NcoI and XhoI restriction sites using the following primers: 5′-GTCGAGATGGTGGAGAAC-3′ (EC34 forward), 5′-AAGATGGGGATCTGG-3′ (EC34 reverse), 5′-GAGAAGTCCGACTACGG-3′ (EC56 forward), 5′-CAGACGAGGGGG-3′ (EC56 reverse). As a template, all of the PCR amplification were performed using the gene of N-terminal FLAG and C-terminal Myc tagged CDH17 (NM_001144663) optimized for codon usage for Sf9 and cloned into a pUC57 vector by GenScript.
EC12 was expressed and purified following the method previously reported.(25,26) Briefly, the expression vector was transformed into E. coli BL21 (DE3) and grown in LB medium containing 50 μg/mL kanamycin at 37°C. Expression was induced by the addition of 0.5 mM IPTG when the optical density at 600 nm reached 0.45 and proceeded at 20°C, overnight. The harvested cells were suspended in buffer A (20 mM Tris, 500 mM NaCl, 3 mM CaCl2, 20 mM imidazole, pH 8.0) and lysed by sonication (UD-201; Tomy, Tokyo). The lysate was centrifuged at 40,000 g for 30 minutes at 4°C. Separation of EC12 from the supernatant was conducted by immobilized metal-ion affinity chromatography (IMAC) using Ni-NTA Agarose (Qiagen). EC12 was eluted with buffer A containing 300 mM imidazole. The protein was treated with His6-Ulp1 protease to remove His6-SUMO at 4°C for 4 h, followed by separation of the His6-tagged proteins by IMAC. Further purification was conducted by size-exclusion chromatography in a HiLoad 26/600 Superdex 200 column (GE Healthcare) using a buffer containing 10 mM HEPES, 150 mM NaCl, and 3 mM CaCl2 at pH 7.5. Expression and purification of EC34 and EC56 were conducted as for EC12 except for the treatment with Ulp1 protease and subsequent IMAC.
ELISA was performed for epitope mapping. Briefly, each of the EC12, 34, and 56 fragment proteins was coated at 1 μg/mL in saline on high-binding polystyrene 96-well plates overnight at 4°C. Then the plates were blocked with reaction buffer for 30 minutes. The plates were incubated with a primary antibody in reaction buffer for 1 hour. After washing thrice with washing buffer (0.05% Tween 20 in saline), the plates were incubated for 1 hour with a HRP-conjugated anti-mouse IgG (Fc fragment-specific) antibody. After washing five times with washing buffer, the enzymatic reaction was visualized with TMB Soluble Reagent. After the reaction was stopped with TMB Stop Buffer, the absorbance was measured at 450 nm using a microplate reader (Biotrak II; GE Healthcare, Piscataway, NJ).
BV-KinExA
BV display assay of Kinetic Exclusion Assay (KinExA) was performed according to a previous report.(27) Budded baculovirus adsorbed magnetic beads (BV-beads) were prepared as follows. Forty milligram of Magnosphere MX200/carboxyl beads were washed thrice with PBS and suspended in 1.5 mL PBS. Then 1.8 mg of BV was added to the suspended beads to 1.2 mg/mL in the final step. The solution was kept rotating with a tube rotator for 16 hours and then the beads were washed thrice with PBS and blocked with reaction buffer. The BV-beads were serially diluted using the reaction buffer. An appropriate concentration of a purified mAb was added into the serially diluted cells or BV-beads. Each solution was incubated at room temperature for 16 hours with rotation. Then the BV-beads were separated with a magnet, and the free mAb present in the supernatant was diluted to an appropriate signal-range concentration with reaction buffer, then measured by KinExA using the anti-mouse IgG (H+L) antibody coated PMMA beads and Alexa Fluor 647-conjugated anti-mouse IgG (H+L) antibody. All of the KinExA experiments were performed in duplicate with a KinExA 3200 system (Sapidyne Instruments, Inc.). The equilibrium dissociation constant (Kd) was obtained using KinExA software and by “n-curve analysis,” which fits all of the given curves to a single Kd value simultaneously.
Immunotoxin cytotoxicity assay
The CCK-8 assay was performed to assess cell viability. Cells were seeded on 96-well plate at 2500 cells per well with a 100 μL of culture medium and incubated overnight at 37°C with 5% CO2. The next day, the serially diluted immunotoxins with culture medium were added on to the cell culture at 10 μL/well. After incubation for 72 hours at 37°C with 5% CO2, 10 μL of CCK-8 solution was added to each well, and the 96-well plate was continuously incubated at 37°C with 5% CO2 for 3 hours, then the absorbance was measured at 450 nm (A450) using a microplate reader (Biotrak II; GE Healthcare, Piscataway, NJ). The assay was performed in triplicate. The cell viability was calculated as follows:
where A450 (experiment) is the absorbance in an immunotoxin added well, A450 (control) is the absorbance in an immunotoxin-free well, and A450 (blank) is the basal absorbance in medium alone.
Combination index calculation
The immunotoxin cocktail effect was evaluated using the combination index (CI) derived from the multiple drug effect equation developed by Chou and Talalay.(28) CalcuSyn software (Biosoft, Cambridge, United Kingdom) was used to calculate the CI values from the immunotoxin-dose dependent fraction affected (Fa) curves. The Fa was calculated from the viability as follows: Fa = 1 − (% viability/100). The cocktail effects were evaluated as CI <1 for synergy, CI = 1 for additive, and CI >1 for antagonism, as suggested.(28) The resulting values were used to construct a plot of CI values over a range of the fractions affected (Fa-CI plot) (Fig. 5B).
Results
Generation and selection of anti-CDH17 mAbs
The human CDH17 gene was inserted into a pFastBac vector with N-terminal FLAG and C-terminal Myc tagged. Recombinant baculovirus harboring the CDH17 gene was generated as described. The expression of CDH17 on BV was confirmed by immunoblotting with an anti-FLAG antibody (Fig. 1A). The immunization with CDH17 expressing BV (CDH17-BV) was performed by a method described previously.(23,24) Briefly, gp64 transgenic mice were immunized with CDH17-BV and then isolated spleen cells were fused with mouse myeloma cells. For screening, cell ELISA was used for the CDH17-expressing gastric cancer cell line AGS as positive and nonexpressing MKN74 cells as the negative control.(13)
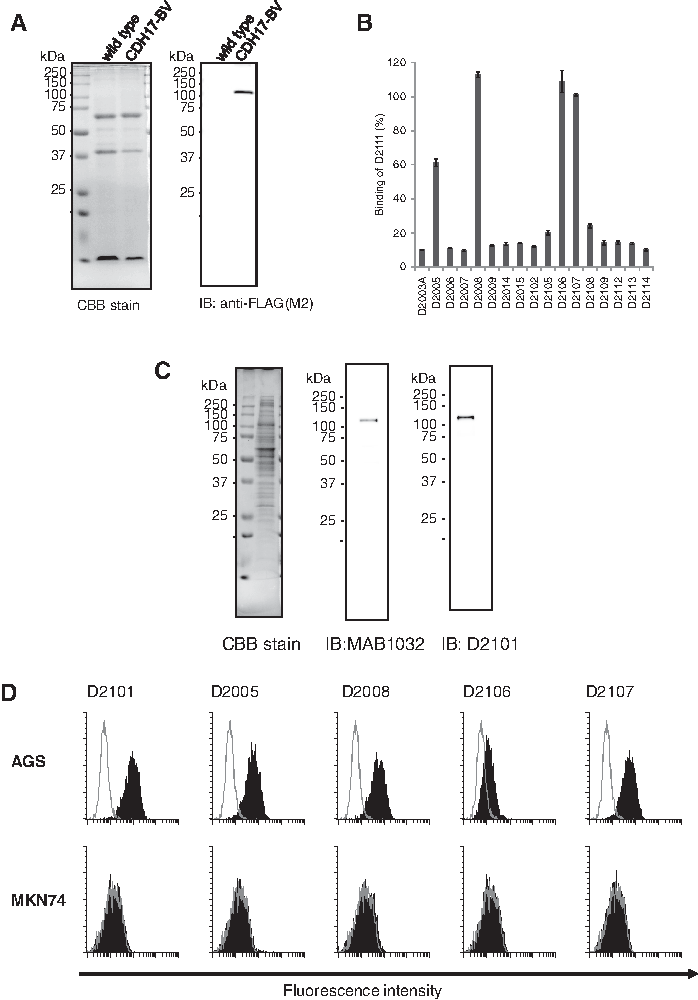
After limiting dilution, we established 22 clones of anti-CDH17 producing hybridomas. Clone D2111 was arbitrarily selected to characterize these mAbs, and a competition assay against D2111 on AGS cells was conducted. The five clones D2001, D2013, D2101, D2103, and D2111 were completely competitive, so classified as recognizing the same epitope (data not shown). The competition assay was further performed using other hybridoma culture supernatants to select the clones recognizing different epitopes (Fig. 1B). Among 17 clones, 4 clones (D2005, D2008, D2106, and D2107) were different from the D2111 epitope. We selected five clones (D2101, D2005, D2008, D2106, and D2107) for further characterization.
Separation of the AGS clones having different CDH17 expression levels
We performed flow cytometry of AGS cells obtained from ATCC using D2101. We found the broad and multipeaked histogram shown in Figure 2. An attempt was made to separate the clones having different CDH17 expression levels by limiting dilution. Consequently, we established three AGS cell lines with different CDH17 expression levels designated as AGSlow, AGSmed, and AGShigh (Fig. 2). The CDH17 expression levels quantified with QIFIKIT (Dako) were 10,000 molecules per cell for AGSlow, 50,000 molecules per cell for AGSmed, and 200,000 molecules per cell for AGShigh. The expression levels of these cell lines were stable for at least 3 months. The AGShigh cell line was used for experiments hereafter unless otherwise stated.

Flow cytometry analysis of CDH17 expression. Filled histogram indicates D2101 applied cells. Open histogram indicates control IgG, B8109B, applied cells. The number of CDH17 proteins per cell is indicated in the top part of each histogram.
MAb characterization
To check the anti-CDH17 mAb specificity, immunoblotting and flow cytometry were performed. For the immunoblot, the commercially available anti-CDH17 mAb (MAB1032; R&D) was used as a reference. As shown in Figure 1C, both MAB1032 and D2101 specifically detected the 120 kDa protein. For flow cytometry, AGShigh and MKN74 cell lines were used as the CDH17 positive and negative control, respectively.(13) As shown in Figure 1D, all five clones exhibited histogram shifts compared to the control antibody (B8109B) in AGS cells, but not MKN74 cells. Thus, these mAbs recognize CDH17 expressing AGS cells specifically.
Round-robin competition assay on AGS cell and binding assay to the fragment of CDH17 proteins was performed to determine the epitope on CDH17 recognized by the anti-CDH17 mAbs. All of the five clones selected above were biotinylated and used at 0.03 μg/mL for D2005, 0.3 μg/mL for D2101 and D2008, and 3 μg/mL for D2106 and D2107 in 50 μg/mL competitive mAb for competition assay on AGS. As shown in Figure 3A, the mAbs did not entirely compete with each other. The binding assay to the fragment of CDH17, EC12, EC34, or EC56 revealed that D2101, D2005, and D2106 recognized EC12 and D2008 recognized EC56 (Fig. 3B, C).
We assessed the dose-dependent binding activity of the mAbs to CDH17 on AGS cells by ELISA (Fig. 4A). The effective concentration at 50% (EC50) values of the mAb binding is shown in Table 1. As the binding activity of D2106 and D2107 was too low, the higher activity clones, D2101, D2005, and D2008, were chosen and then BV-KinExA was applied to them to determine the equilibrium dissociation constant (Kd). Both D2101 and D2005 displayed a Kd of 1.6 nM with considerable accuracy. Because the suitable Kd range for KinExA is from 10−12 to 10−8 M,(29) D2008 displayed that a Kd of 138 nM has a broad confidence interval (Table 1 and Fig. 4E).

All data shown are the means ± standard deviations.
95% Confidence intervals are indicated in parentheses.
BV, budded baculovirus; EC50, effective concentration at 50%; ELISA, enzyme linked immunosorbent assay; mAb, monoclonal antibody.
Immunotoxin cytotoxicity
To demonstrate the cytotoxic activity of these mAbs to the AGShigh group, each biotinylated mAb was conjugated to streptavidin-saporin to generate the immunotoxins. Then each immunotoxin (IT) that had been diluted serially from 4.2 nM was applied on the AGS cells. The EC50 cytotoxicity values were 18 pM for D2101IT, 23 pM for D2005IT, and 330 pM for D2008IT (Fig. 4C and Table 1). As shown in Figure 4D, the cytotoxic activity of D2101IT depended on the density of CDH17 on the cell.
Enhanced immunotoxin cocktail cytotoxicity
To determine whether the individual mAbs in the cocktail bind to CDH17 additively, we checked the binding activity of various combinations of mAbs to AGShigh by cell ELISA (Fig. 4B). The binding of each mAb reached saturation at ∼1 μg/mL. As shown in Figure 4B, the binding signal of two mAb combined cocktails increased more compared with each mAb alone at the saturated concentration (D2005 + D2008 > D2005 or D2008, D2101 + D2005 > D2101 or D2005, D2101 + D2008 > D2101 or D2008). The combination of three mAbs displayed the maximum signal among all tested.
Next, the immunotoxin cocktail of D2101IT, D2005IT, and D2008IT mixed at a 1:1:1 ratio was applied to the three different CDH17 levels of AGS cell lines (Fig. 5A). The immunotoxin cocktail exhibited significant cytotoxic effect compared to the D2101IT alone (p < 0.0001 for AGSlow, p < 0.001 for AGSmed, and p < 0.01 for AGShigh).

The multiple drug effects of the immunotoxin cocktail were analyzed with CalcuSyn software to determine the CI values. As each immunotoxin individually exhibited low cytotoxic activity for the AGSlow, accurate CI values for AGSlow were not obtained. Thus, the CI value calculation was applied to the AGShigh and AGSmed. Figure 5B shows a Fa-CI plot of the immunotoxin cocktail against AGShigh or AGSmed. In the AGSmed group, powerful synergistic effect (CI <1) was observed at Fa >0.5. While in the AGShigh group, additive or moderate antagonism (CI = 1–1.2) was observed at Fa >0.2.
Discussion
CDH17 has been shown to be a useful diagnostic marker of gastric cancer and hepatocellular carcinoma (HCC).(6,7,9) Previous studies demonstrated CDH17 to be a potential therapeutic target; anti-CDH17 mAb binding or CDH17 knockdown inhibits cancer cell growth.(8,12–15) However, at present the targeting CDH17 for ADC or immunotoxin therapy has not been reported. We generated five anti-CDH17 mAbs recognizing different epitopes on CDH17 to develop a therapeutic agent targeting CDH17 (Fig. 3). The anti-CDH17 mAbs (D2101, D2005, and D2008) conjugated indirectly with saporin displayed cytotoxic activity against AGS cells expressing a high level of CDH17 (200,000 antigens per cell) (Fig. 4C). In a comparison of the cytotoxicity of D2101IT against AGS cells with different CDH17 levels, it exhibited poor cytotoxic activity to AGS expressing low level CDH17 (10,000 antigens per cell) (Fig. 4D). These results suggest that the cytotoxic activity by the anti-CDH17 immunotoxin depends on the CDH17 antigen density on the cell.
As cancer cells possess considerable diversity, the expression level of antigens on a cell is not homogeneous.(30) For the effective treatment of tumors, a therapeutic strategy against the cancer cells expressing a low level of CDH17 is desirable. Toward this goal, mAb cocktail therapy is one of the promising strategies. For example, cocktails of naked mAb have been shown to exert synergistic or additive effect against not only infectious diseases for the purpose of viral neutralization but also in cancer cells for cytotoxicity using antibody-dependent cell-mediated cytotoxicity (ADCC), and complement dependent cytotoxicity (CDC).(31) Moreover, cocktails of immunotoxins or radiolabeled mAbs recognizing different antigens have been used to increase the number of binding site per cell to improve antitumor efficacy.(20–22,32)
In this study, it was found that the cocktail of immunotoxins targeting different epitopes on a single molecule, CDH17, displayed promising activity in terms of additive or synergistic effect. The immunotoxin cocktail targeting distinct epitopes of CDH17 exerted a significant effect against a gastric cancer cell line AGS expressing a lower level CDH17 (Fig. 5A). The multidrug effect evaluation by Chou and Talalay(28) showed the effect to be synergistic by means of an evaluation of the CI value, which is clear in the AGSmed case. Although the mechanism which confers the synergy is unclear, the immunotoxin cocktail has the potential to facilitate CDH17 internalization for low-level CDH17-expressing tumor cells. The induction of internalization by the clustering of antibody receptor of HER2 has been previously reported.(33,34) The key mechanism of action for the immunotoxin cocktail effect observed in this study is hypothesized to be the use of multiple antibodies that bind to distinct epitopes of CDH17 responsible for additional linkages between surface CDH17 molecules. That may induce CDH17 clustering, which enhances internalization and results in a greater amount of toxin delivered into the target cells. The cytotoxic activity of ADC is generally correlated with the level of target antigen expression on the cell surface.(34–36) This suggests that the therapeutic effect needs to enhance killing of not only high level but also low level target antigen expression on the cell surface. In this respect, the immunotoxin cocktail targeting distinct epitopes provides a promising effect for cancer therapy. Lin et al. reported that over 60% of their tested 30 panel gastric cancer cell lines are CDH17 positive, and nearly half of these cell lines have lower CDH17 expression.(13) As the immunotoxin cocktail method is useful for the low CDH17 expression level of AGS cell line in our study, more than 60% of gastric cancer cell lines may show a cytotoxicity by this method. This cocktail therapy would be applicable when the target protein comprises multiple domains.
In the future, as reported in chemotherapy,(37) even the combination of immunotoxins and/or ADCs with different mechanisms of action in antibody cocktail will increase cytotoxic activities and decrease side effects.
Footnotes
Acknowledgments
The authors thank Dr. Kevin Boru of Pacific Edit for review of the article. This work was supported by the Program for Development of New Functional Antibody Technologies of the New Energy and Industrial Technology Development Organization (NEDO) of Japan.
Author Disclosure Statement
No competing financial interests exist.