
Abstract
The highly O-glycosylated membrane glycoprotein podoplanin (PDPN) is frequently overexpressed in several malignant cancers, such as oral cancer, lung cancer, germinal neoplasia, mesothelioma, and brain tumor. The expression of PDPN is strongly associated with cancer progression and poor prognosis. PDPN possesses three tandem repeats of platelet aggregation-stimulating (PLAG) domains (PLAG1, PLAG2, and PLAG3) and PLAG-like domain (PLD), and binds to C-type lectin-like receptor 2 (CLEC-2) on platelets, followed by PDPN-mediated platelet aggregation. We have previously established a novel anti-Tasmanian devil PDPN (tasPDPN) monoclonal antibody (mAb), PMab-233, which specifically detects tasPDPN using flow cytometry, Western blot, and immunohistochemical analyses. However, the specific binding epitope of tasPDPN for PMab-233 remains to be clarified. Herein, a series of deletion or point mutants of tasPDPN were utilized for investigating the binding epitopes of PMab-233 using flow cytometry. The findings of this study demonstrated that Asp30, Thr33, and Thr34 of tasPDPN, which are located in PLAG1, are responsible for the binding of PMab-233 to tasPDPN.
Introduction
Podoplanin (PDPN)/Aggrus/T1α is a highly O-glycosylated transmembrane glycoprotein, and is extensively distributed in normal tissues, such as podocytes of the kidneys, type I alveolar cells of the lungs, lymphatic endothelial cells of all organs, myofibroblasts, mesothelial cells, and central nervous system.(1–4) Many physiological functions of PDPN have been reported to play crucial roles in blood/lymphatic vessel separation,(5,6) embryonic cardiac development,(7,8) and high endothelial venule integrity.(9)
The PDPN overexpression has also been observed in several malignant tumors, such as brain tumors,(10,11) oral cancers,(12) lung cancers,(13) melanomas,(14) mesotheliomas,(15) breast cancers,(16,17) and osteosarcomas.(18–20) Clinical data demonstrated that PDPN expression is associated with poor prognosis and tumor malignancy in lung carcinomas, oral squamous cell carcinomas, and breast cancers.(8,21–24) PDPN facilitates hematogenous metastasis by eliciting tumor cell-induced platelet aggregation response through its interaction with platelet C-type lectin-like receptor 2 (CLEC-2).(25–28) These pieces of evidence imply the importance of developing anti-PDPN monoclonal antibodies (mAbs) for cancer therapeutic treatment.
We have recently established a novel PMab-233 (IgG1, kappa), which specifically detects Tasmanian devil PDPN (tasPDPN) using Cell-Based Immunization and Screening (CBIS) method.(29) PMab-233 detected Chinese hamster ovary (CHO)/tasPDPN cells by flow cytometry and recognized tasPDPN protein by Western blotting. Furthermore, PMab-233 strongly detected CHO/tasPDPN cells by immunohistochemistry. These findings suggest that PMab-233 may be useful as a lymphatic endothelial cell marker of the Tasmanian devil. However, the specific binding region of tasPDPN for PMab-233 remains to be elucidated. In this study, we investigated the binding epitopes of PMab-233 by analyzing a series of deletion or point mutants of tasPDPN using flow cytometry.
Materials and Methods
Cell lines
CHO-K1 was purchased from the American Type Culture Collection (ATCC, Manassas, VA). The tasPDPN mutation plasmids containing PA16 tag were transfected into CHO-K1 cells using Lipofectamine LTX (Thermo Fisher Scientific, Inc., Waltham, MA). Transiently transfected cells with deletion/point mutants were cultured in RPMI 1640 medium (Nacalai Tesque, Inc., Kyoto, Japan), supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.), 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 25 μg/mL of amphotericin B (Nacalai Tesque, Inc.) at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Production of tasPDPN mutants
The synthesized DNA of tasPDPN was subcloned into a pCAG vector (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan), and PA16 tag (GLEGGVAMPGAEDDVV) was added at the N-terminus. The deletion mutants of tasPDPN produced using PCR were subcloned into pCAG vector with PA16 tag using the In-Fusion HD Cloning Kit (Takara Bio, Inc., Shiga, Japan). Substitutions of amino acids to alanine in tasPDPN sequence were conducted using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Inc., Santa Clara, CA). These constructs were verified using direct DNA sequencing.
Flow cytometry
Transiently transfected CHO-K1 cells were detached by 0.25% trypsin/1 mM ethylenediaminetetraacetic acid (EDTA; Nacalai Tesque, Inc.). After washing with 0.1% bovine serum albumin (BSA)/phosphate buffered saline (PBS), the cells were incubated with anti-tasPDPN antibody (PMab-233; 1 μg/mL) or control anti-PA16 tag antibody (NZ-1; 1 μg/mL) for 30 minutes at 4°C followed by treatment with Alexa Fluor 488-conjugated antimouse IgG (1:1000; Cell Signaling Technology, Inc., Danvers, MA) and Oregon Green-conjugated antirat IgG (1:1000; Thermo Fisher Scientific, Inc.), respectively. Fluorescence data were collected using a Cell Analyzer EC800 (Sony Corp., Tokyo, Japan).
Results and Discussion
In our previous study, we established a novel anti-tasPDPN mAb, PMab-233, which can be efficiently utilized for flow cytometry, Western blotting, and immunohistochemical detection of tasPDPN.(29) Unfortunately, we could not investigate the PDPN expression using normal tissues of Tasmanian devil; therefore, we used formalin-fixed paraffin-embedded CHO/tasPDPN cells. PMab-233 did not react with human, mouse, rat, rabbit, dog, bovine, cat, pig, horse, tiger, alpaca, bear, goat, sheep, or whale PDPNs, which indicates that PMab-233 is specific to tasPDPN.(29) On the basis of these results, the epitope mapping of PMab-233 could be beneficial in uncovering the pathophysiological function of tasPDPN.
Although three tandem repeats of platelet aggregation-stimulating (PLAG) domain (PLAG1, PLAG2, PLAG3) are observed in human or mouse PDPNs,(30) PLAG2 and PLAG3 do not exist in tasPDPN (Fig. 1A). In contrast, two PLAG-like domains were observed. We first constructed six deletion mutants of tasPDPN (Fig. 1A). Transient transfections of tasPDPN-mutant clones were produced using CHO-K1 cells, including dN30 (corresponding to 30–143 amino acids [aa]); dN40 (corresponding to 40–143 aa); dN50 (corresponding to 50–143 aa); dN60 (corresponding to 60–143 aa); dN70 (corresponding to 70–143 aa); and dN80 (corresponding to 80–143 aa). All deletion mutants of tasPDPN contain N-terminal PA16 tag and were analyzed using flow cytometry for epitope mapping of PMab-233. NZ-1 (anti-PA16 tag mAb) detected all deletion mutants of tasPDPN, including dN30, dN40, dN50, dN60, dN70, and dN80 (Fig. 1B). In contrast, PMab-233 lost the reaction with dN30, dN40, dN50, dN60, dN70, and dN80 (Fig. 1C). These results imply that N-terminus of the epitope-binding region of PMab-233 is located between the 27th and 30th amino acids.

Epitope mapping of PMab-233 using deletion mutants of tasPDPN.
Next, we investigated the epitope-binding region of PMab-233 by producing 10 point mutants of tasPDPN, including L27A, P28A, E29A, D30A, A31G, A32G, T33A, T34A, I35A, and D36A. All these mutants can be recognized by NZ-1 (Fig. 2A). We observed that PMab-233 reacted with L27A, P28A, E29A, A31G, A32G, I35A, and D36A mutants, and weakly reacted with D30A using flow cytometry (Fig. 2B). In contrast, PMab-233 did not react with T33A and T34A (Fig. 2B).
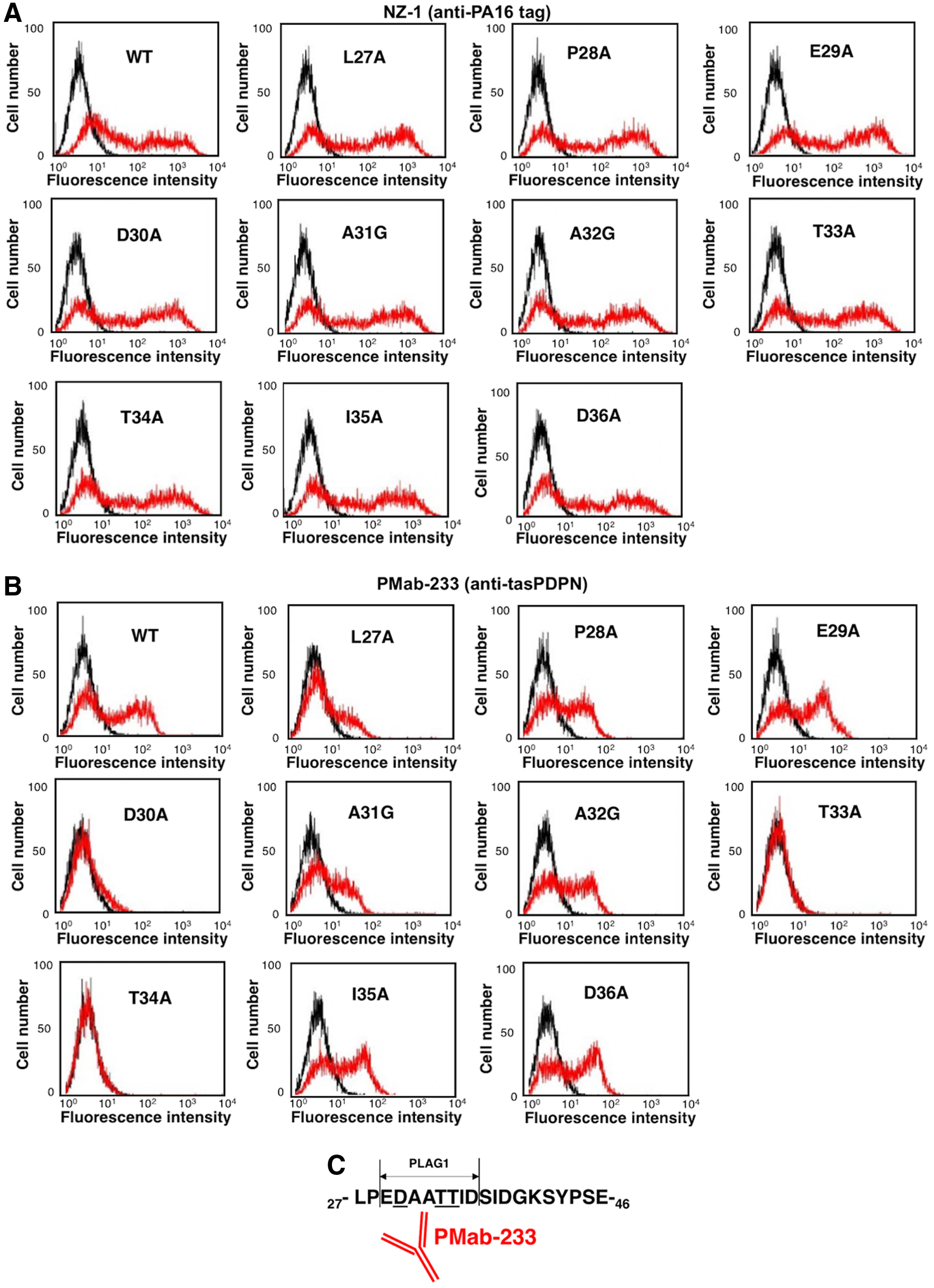
Epitope mapping of PMab-233 using point mutants of tasPDPN.
In conclusion, we demonstrated that Asp30, Thr33, and Thr34 of tasPDPN are critical for PMab-233-specific binding to tasPDPN (Fig. 2C). Asp30, Thr33, and Thr34 are located in PLAG1 domain of tasPDPN. PMab-233 can be a useful tool in elucidating the pathophysiological function of tasPDPN.
Footnotes
Author Disclosure Statement
The authors have no conflict of interest.
Funding Information
This research was supported, in part, by AMED under Grant Numbers JP19am0401013 (Y.K.), JP19am0101078 (Y.K.), and JP19ae0101028 (Y.K.), and by JSPS KAKENHI Grant Number 17K07299 (M.K.K.) and Grant Number 19K07705 (Y.K.).