
Abstract
The human norovirus (HuNov) major capsid VP1comprises an S (shell) and a P (protruding) domain; the latter is responsible for virus attachment and infection. The dimeric formation of P (containing P1 and P2 subdomains) is indispensable for forming a receptor-binding pocket, enabling HuNov to dock to attachment factor histo-blood group antigens (HBGAs) on the host cell. Thus, the P-specific antibody may hamper the engagement of P and HBGA, thereby inhibiting virus infection. In this study, we developed and characterized two HuNov P-specific murine monoclonal antibodies (MAbs), namely, 5C6 and 1H12. They can bind to P protein with high affinity, as evidenced by the results of indirect fluorescent assay, western blot, and Biolayer interferometry assay. Particularly, the MAb 1H12 recognizes the P2 subdomain, whereas the 5C6 targets the distal P1. These MAbs may contribute to the exploration of novel epitopes on HuNov VP1 and to the development of new antivirals.
Introduction
Human norovirus (HuNov) is a leading cause of acute gastroenteritis, manifesting severe clinical syndromes, including diarrhea, vomiting, fever, and abdominal pain, in subjects of any age. 1 The HuNov genome is approximately 7.5 kb in length and encodes six nonstructural and two structural proteins, i.e., the major capsid protein VP1 (∼ 59 kDa) and the minor capsid protein VP2 (∼ 29 kDa). 2 The viral capsid is composed of 90 dimers of VP1, which is divided into a shell (S) domain and a protruding (P) domain. 3 The P domain, responsible for viral attachment to host cells through binding to bile acids, histo-blood group antigens (HBGAs), cations, and sialic acid, 4 is therefore proposed as the most important target during antiviral reagent development against HuNov.
So far, the HuNov-specific neutralizing antibodies revealed can be mainly grouped into three categories given hereunder.5–8 First, antibodies competitively binding to the receptor-binding pocket of HuNov VP1 directly inhibit the attachment of HuNov to host cells. 9 Second, antibodies binding to epitopes proximal to the receptor-binding pocket occlude virus infection through steric clashes against HBGA. 10 Third, antibodies binding to epitopes far away from the receptor-binding pocket deprive HuNov infectivity by improving disassembly or degradation of HuNov particles. 11
Nevertheless, monoclonal antibodies (MAbs) targeting novel epitopes on HuNov virions are still worthy of further exploration. In this current study, we developed and characterized two murine MAbs, targeting distinct subdomains of the HuNov P protein.
Materials and Methods
Preparation of MAbs against HuNov P
The coding sequence for the P domain of a GII.4 strain (GenBank accession number LC699526) was synthesized (Tsingke, Beijing, China) and cloned into the pET28a expression vector (pET28a-p). The recombinant plasmid was then transferred into BL21 competent cells. The P protein was successfully expressed in the BL21 harboring pET28a-p upon the induction at 0.5 mM isopropyl β-D-thiogalactoside (IPTG). The preparation of His-tagged HuNov P and its corresponding murine MAbs were described previously. 12 Briefly, five 6-week-old female BALB/c mice were subcutaneously immunized three times, two weeks apart, with 100 μg of the HuNov P adjuvanted with Freund’s adjuvant. Mice splenocytes were harvested and fused with SP2/0 cells using 50% polyethyleneglycol (Sigma-Aldrich, MO). HuNov P-specific antibodies in culture supernatants of the fused cells were screened using enzyme-linked immunosorbent assay (ELISA). The positive hybridoma cells were repeatedly cloned by a limited dilution, and the stable hybridoma clones were injected into abdominal cavities of BALB/c mice to yield sufficient antibodies. Subsequently, the MAbs were harvested from the seroperitoneum and purified using an antibody purification kit according to the manufacturer’s specifications (NAbTM Protein A/G Spin Kit, Thermo Scientific, USA). The animal study was approved by the ethics committee of the Wuhan University of Bioengineering, China (permit number: WUB20230124).
Immunofluorescence assay
HEK293T cells were seeded into 24-well tissue culture plates (Costar Corning Inc., NY, USA) at a density of 1 × 105 cells per well in a 0.5 mL RPMI 1640 complete medium. When the cells reached approximately 70% confluence, the culture medium was removed, and then, cells were washed three times with phosphate buffered saline (PBS [pH7.4]) and transfected by plasmid pCAGGS-p for 20 hours at 37°C. The transfected cells were fixed with absolute methanol and incubated with MAbs for 2 hours, followed by an application of fluorescein isocyanate-conjugated goat antimouse IgG. Fluorescent images were examined under a fluorescent microscope.
Immunoblotting
After washing twice with PBS the transfected HEK293T cells were harvested, centrifuged to remove the supernatant, and then lysed with cell lysis buffer supplemented with protease inhibitor cocktail (Roche). Cleared cell lysate supernatants were separated by 10% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then electrophoretically transferred to nitrocellulose transfer membrane (GE Healthcare, Darmstadt, Germany). The membrane was blocked for 1 hour at room temperature (RT) with blocking solutions containing 1% bovine serum albumin in TBS (20 mM Tris-HCl [pH7.5], 150 mM NaCl), and then incubated with MAbs at a concentration of 1 μg/mL overnight at 4°C (or 2 hours at RT). After washing five times with T-TBS (20 mM Tris-HCl [pH7.5], 150 mM NaCl, 0.05% Tween 20), the membrane was incubated for 30 m with HRP-conjugated goat antimouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). After washing three times with T-TBS, the membrane was developed by treatment with ECL Western Blotting Detection Reagents (GE Healthcare, Darmstadt, Germany).
ELISA
Ninety-six-well ELISA plates were coated with 100 μL of indicated antigen (2 μg/mL of P protein, peptide, or norovirus GII.4 VLP [ab256447, Abcam]) in coating buffer (50 mM NaHCO3, 50 mM Na2CO3 [pH 9.6]) at 4°C overnight. The serially diluted antibody was incubated in the plates overnight at 4°C (or 2 hours at RT). MAbs and irrelative MAb 5G10 (a salmonella bacteria flagellin-specific MAb) 13 were detected using alkaline phosphatase (AP)-labeled goat antimouse IgG (SouthernBiotech, AL, USA), followed by substrate p-nitrophenyl phosphate (Sigma-Aldrich, MO, USA). The reciprocal values of the last dilution giving an OD405nm reading that was twice the background of the empty wells were determined as positive.
Dot blot
Dot blot assay was performed as described previously. 14 Different amounts of P protein (100, 10 1, 0.1, or 0.01 ng) were dropped onto a nitrocellulose membrane; then, the membrane was blocked for 1 hour at RT. The nitrocellulose membrane hybridized with MAbs (1 μg/mL) for 1 hour at RT. After washing five times with T-TBS, the membrane was incubated for 30 minutes with HRP-conjugated antimouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). After washing three times with T-TBS, the membrane was developed by treatment with ECL Western Blotting Detection Reagents (GE Healthcare, Darmstadt, Germany).
Biolayer interferometry assay
Binding assays were performed in 96-well microplates by Octet Red system (Ferbio). First, APS sensors were rinsed in PBS. Second, APS sensors were coupled with 200 μL PBS with MAbs (1 μg/mL). Third, APS sensors were moved into PBS and incubated to clear unabsorbed MAbs. Lastly, APS sensors were exposed to P at concentrations of 1 μg/mL. Association was monitored for 1800 seconds followed by dissociation in PBS alone for another 1800 seconds. The standard curve was measured at the beginning and end of the assay to confirm that it was reproducible and valid over the time taken to run all rows of samples. Data were processed automatically using the Octet User Software version 3.1.
ELISA additive tests
The additive test analysis was performed as described previously. 15 A 96-well ELISA plate was coated with 100 μL of the indicated antigen (0.01 μg/mL of P protein) in coating buffer at 4°C overnight. Additive index (AI) = [2 × A1 + 2 / (A1 + A2) − 1] × 100, where A1 (or A2) was the OD405nm obtained when an MAb was assayed individually, and A1 + 2 was the OD405nm when two MAbs were applied together. The AI would be negligible if both MAbs detected target toward the same epitope or at two adjacent epitopes. In contrast, the AI would be close to 100 when the two epitopes were topographically unrelated. The lowest AI reported for MAbs at different epitopes on P was considered as the threshold for evaluating epitopic correlation.
Results and Discussion
Preparation of HuNov P protein
In order to obtain HuNov P protein, a recombinant plasmid containing HuNov gene fragment encoding P protein (GII.4, LC699526) was constructed and designated pET28a-p. As shown in Figure 1A, a unique protein band emerged with a molecule mass of about 38 kDa (Lane 2) that was in agreement with the anticipated molecule weight of P (305 amino acids, AA). To further confirm whether the protein weighing about 38 kDa mentioned above was P, we utilized anti-Histag (Proteintech, CN) to immunoblot with induced BL21 upon IPTG by western blotting (WB). As seen in Figure 1B, the induced BL21 containing pET28a-p plasmid generated a single specific protein band (∼38 kDa) (Lane 2), which was in agreement with the result of SDS-PAGE. Subsequently, the induced target protein (P) was then purified using affinity chromatography Ni resin (GE healthcare, Darmstadt, Germany) after mass culture of BL21 containing pET28a-p. Collectively, the P protein was induced and produced with high purity. In turn, the P protein was used to immunize five BALB/c mice.

Expression of prokaryotic HuNov P protein.
Characterization of MAbs targeting HuNov P
Two stable positive hybridoma cell lines, designated 1H12 and 5C6, were established. The characteristics of MAbs 1H12 and 5C6 were determined by indirect fluorescent assay (IFA) and WB. Due to the lack of a convenient in vitro culture system for HuNov, we harnessed recombinant plasmid pCAGGS-p expressing eukaryotic P to evaluate the affinity between MAbs and P. Prior to the current study, we have already utilized commercial antibody (2002-G5, Abcam) to validate our experimental system. (Data not shown) An IFA was performed to determine the binding activity of the obtained MAbs after plasmid pCAGGS-p was transfected to HEK293T cell for 20 hours. The antisera from P-immunized mice acted as positive antibody control. As shown in Figure 2A, a fluorescent signal can be seen when 1H12 and 5C6, but not 5G10 (a MAb against bacterial flagellin) 13 were applied as detection antibodies. Of note, the P protein was located predominantly in the nucleus of HEK293T at this experimental circumstance. In addition, we also found P protein used in this assay specifically located in the nucleus of three other cell types, such as Hela, PK15, and Caco2 (data not shown). Moreover, the binding activity of 1H12 and 5C6 to P was evaluated by immunoblotting. The plasmid transfected cells were subjected to WB. As seen in Figure 2B, both 1H12 and 5C6 can specifically interact with capsid P (about 43 kDa) as well. Notably, the P protein expressed in HEK293T is much larger than that expressed in BL21 (about 38 kDa), which was likely due to modifications for P in the eukaryotic expression system, such as glycosylation or phosphorylation.
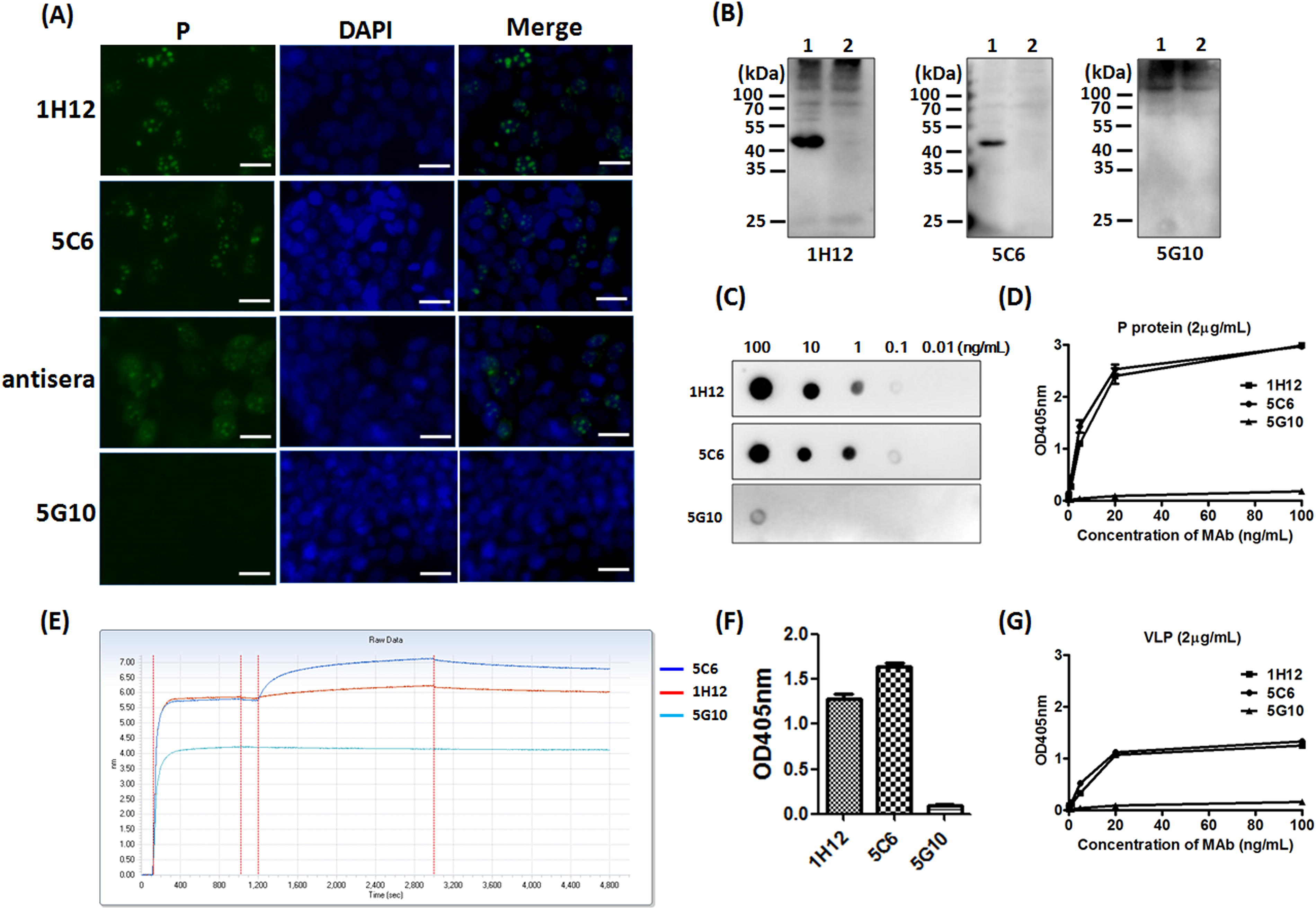
Characterization of two MAbs targeting HuNov P.
The affinities of 1H12 and 5C6 against P were assessed by dot blot. One μL of P protein was dropped onto the nitrocellulose membrane with a concentration ranging from 100 to 0.01 ng/mL. Both MAbs 1H12 and 5C6 can detect P protein as low as 0.1 ng/mL (Fig. 2C). Furthermore, the affinities of 1H12 and 5C6 against P were assessed by ELISA. As seen in Figure 2D, the affinity dynamics showed that 5C6 had slightly stronger binding capacity to P protein compared with 1H12. The ELISA result revealed that both MAbs 1H12 and 5C6 belong to IgG1 subtype (data not shown). In addition, the affinities between P protein and MAbs were tested by the BLI assay. The result show that MAbs 5C6 and 1H12 can bind P protein with affinity constants of 16.78 and 21.22 nM, respectively. (Fig. 2E) Moreover, norovirus VLP was used to detect the binding capacities of the MAbs to virions by ELISA. As observed in Figure 2F and 2G, both MAbs 5C6 and 1H12 can bind to norovirus VLP. Nevertheless, we noticed that the OD values decrease to 1.45 when VLP serve as capture protein, whereas P proteins generate OD values to 3.0. (Fig. 2D) Therefore the MAb affinities to VLP are significantly lower than that to P protein. We believed that the application of prokaryotic P protein during MAb generation resulted in stronger affinity of MAbs to conformational or linear epitopes on P protein compared with that on VLP. The MAbs in this study may not be completely matched with the corresponding epitopes on norovirus VLP, due likely to possible modifications or tiny conformational changes on virions. Therefore, both 5C6 and 1H12 developed in this study can specifically bind to HuNov virion, albeit with lower affinity compared with P protein.
Two MAbs target distinct subdomains of P
An additive assay was performed to judge the position relationship of epitopes recognized by 1H12 and 5C6. AI between 1H12 and 5C6 is about 70, which indicates that the epitopes recognized by 1H12 and 5C6, respectively, are totally separated on the P protein. (Fig. 3A) Based on the crystal structure (PDB: 5F4J), the HuNov protruding domain (P) is composed of two parts (P1 and P2 subdomains) (Fig. 3B). Notably, P1 contains N terminal 53AA and 125 AA at C terminus of P, whereas P2 only includes 127AA in the middle of P (Fig. 3C). The recombinant plasmid pCAGGS-p1 was constructed by overlapping PCR (the primers for the upstream fragment were GT
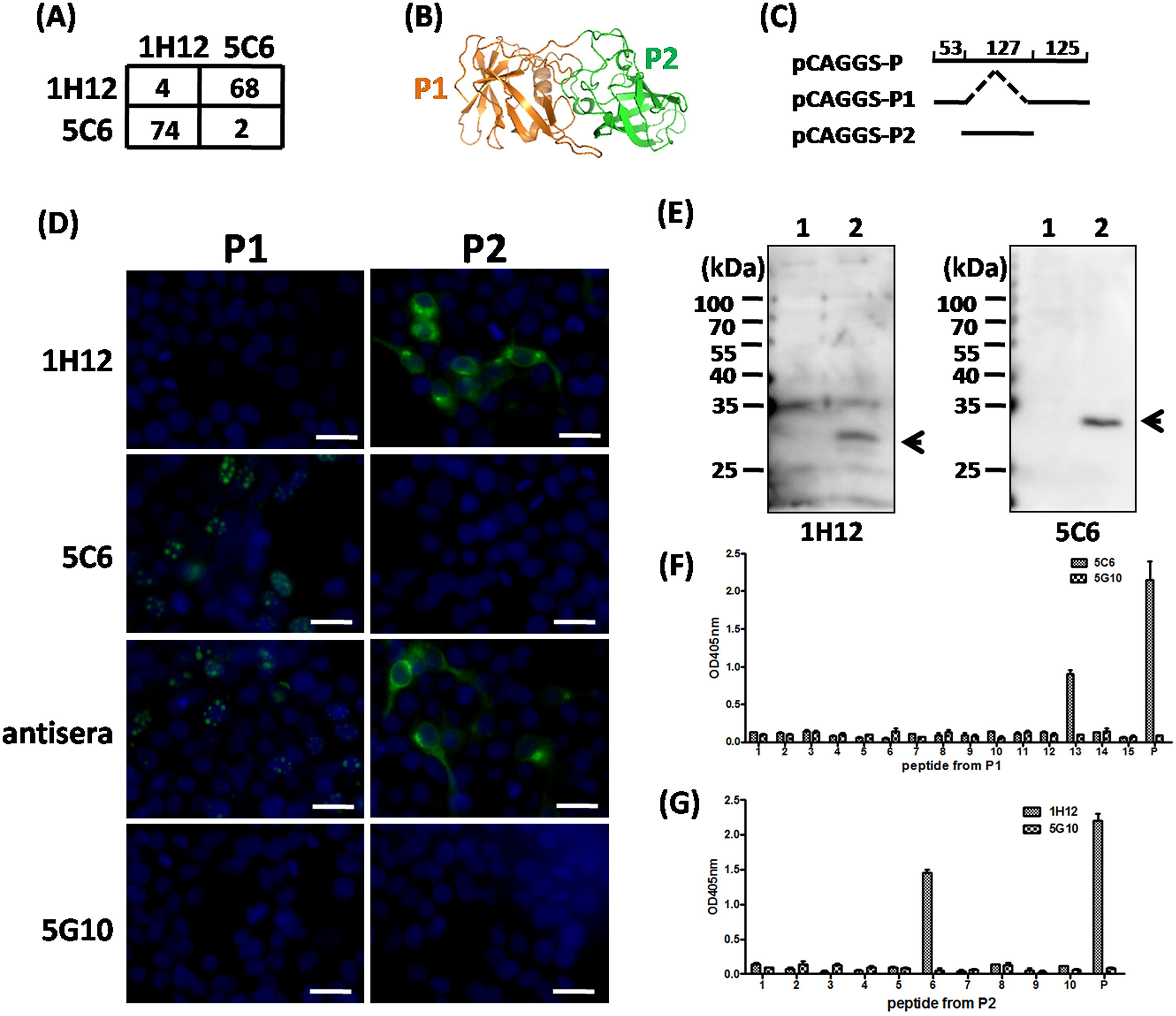
MAbs 1H12 and 5C6 recognized two distinct epitopes on P.
Footnotes
Acknowledgments
The authors sincerely thank Dr. Shiping Wu at the Wuhan University of Bioengineering for editorial assistance.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the Hubei Provincial Natural Science Foundation of China (2020CFB520) and High-level Scientific Research Foundation for the introduction of talent of Wuhan University of Bioengineering (2017KQ01).