
Abstract
The C-C motif chemokine receptor 8 (CCR8) is highly and selectively expressed in regulatory T (Treg) cells and is associated with tumor progression. The massive accumulation of Treg cells into tumors suppresses the effector function of CD8+ cells against tumor cells. Therefore, selective depletion of Treg cells using anti-CCR8 monoclonal antibodies (mAbs) reinvigorates antitumor immune responses and improves responses to cancer immunotherapy. Previously, we developed an anti-mouse CCR8 (mCCR8) mAb, C8Mab-2, using the Cell-Based Immunization and Screening method. In this study, the binding epitope of C8Mab-2 was investigated using flow cytometry. The mCCR8 extracellular domain-substituted mutant analysis showed that C8Mab-2 recognizes the N-terminal region (1–33 amino acids) of mCCR8. Next, 1×alanine (or glycine) scanning and 2×alanine (or glycine) scanning were conducted in the N-terminal region. The results revealed that the 17-DFFTAP-22 sequence is important for the recognition by C8Mab-2, and Thr20 is a central amino acid of the epitope. These results revealed the involvement of the N-terminus of mCCR8 in the recognition by C8Mab-2.
Introduction
The C-C motif chemokine receptor 8 (CCR8) is a member of G protein-coupled receptors (GPCRs) family. The C-C motif chemokine ligands (CCLs), including CCL1, CCL8, CCL16, and CCL18, are known as ligands for human CCR8. CCL1 is the only ligand for CCR8, 1 which is produced by CD11b+CD14+ myeloid cells during the infiltration of regulatory T (Treg) cells into tumor. 2 Upon binding of CCL1 to CCR8, the FOXP3 is upregulated by the STAT3 pathway, and the activated CCR8+ Treg cells potently suppress antitumor immunity through secretion of granzyme B and IL‐10. 3 Increased expression of CCR8 is observed in Treg cells, especially in cancer patients. 4 Patients with high levels of Treg cells exhibit poor prognoses and clinical outcomes in several cancers. 5 Therefore, it has been proposed that depletion of tumor-infiltrated Treg cells could restore antitumor immunity and improve responses to tumor immunotherapy. 6 Recent preclinical mouse models have revealed that depletion of Treg cells using an anti-mouse CCR8 (mCCR8) monoclonal antibody (mAb) exhibited strong antitumor responses through dramatic changes of the intratumor CD8+ T-cell profile 7 or enhanced the antitumor effects of antiprogrammed cell death 1 (PD-1) therapy. 8
The understanding of the structural-based CCR activation is important for the development of therapeutic agents. Among the CCR family members, CCR2 and CCR5 have been structurally solved in both inactive and active states,9–12 while inactive state of CCR7 and CCR9 and active state of CCR1 and CCR6 structures are also characterized.13–16 Furthermore, the structures of CCR8 in complex with either the antagonistic mAb or the endogenous ligand CCL1 were determined, which provides the specific activation mechanism by CCL1 and inhibition by mAb. 17
We have developed anti-mouse GPCR mAbs against CCR1 (clone C1Mab-6), 18 CCR3 (clones C3Mab-2, C3Mab-3, and C3Mab-4),19–21 CCR8 (clones C8Mab-1, C8Mab-2, and C8Mab-3),22–24 CXCR1 (clone Cx1Mab-1), 25 CXCR3 (clone Cx3Mab-4), 26 and CXCR4 (clone Cx4Mab-1) 27 using the Cell-Based Immunization and Screening (CBIS) method. For the determination of the epitopes, we have faced difficulty using conventional methods such as enzyme-linked immunosorbent assay. In this study, epitope mapping of an anti-mCCR8 mAb was conducted by flow cytometry-based approaches.
Materials and Methods
Cell lines
Chinese hamster ovary (CHO)-K1 cell was obtained from the America Type Culture Collection (ATCC, Manassas, VA). Stable transfectants of CHO/mCCR8 were established in our previous study. 24 Briefly, synthesized DNA (Eurofins Genomics KK, Tokyo, Japan) encoding mCCR8 (Accession No.: NM_007720.2) was subcloned into a pCAG-Ble vector (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). mCCR8 plasmid was transfected using a Neon Transfection System (Thermo Fisher Scientific Inc., Waltham, MA). CHO/mCCR8 cells were sorted using a cell sorter (SH800; Sony Corp., Tokyo, Japan) and cultivated in a medium containing 0.5 mg/mL of Zeocin (InvivoGen, San Diego, CA).
The chimeric and the point mutant plasmids were transfected into CHO-K1 cells using the Neon Transfection System (Thermo Fisher Scientific Inc.). Stable transfectants were selected using SH800. Cells were cultured in Roswell Park Memorial Institute−1640 medium (Nacalai Tesque, Inc., Kyoto, Japan) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific Inc.), 100 units/mL of penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (Nacalai Tesque, Inc.) at 37°C in a humidified atmosphere containing 5% CO2. The stable transfectants were cultivated in a medium containing 0.5 mg/mL of Zeocin.
Plasmid construction
Synthesized DNAs (Eurofins Genomics KK) encoding mouse CCR3 (mCCR3; Accession No.: NM_009914.4) and mCCR8 were subcloned into a pCAG-Ble vector. Chimeric mutants including mCCR3 (mCCR8p1–33), mCCR3 (mCCR8p92–105), mCCR3 (mCCR8p170–200), and mCCR3 (mCCR8p262–278) were produced with a PA16 tag at their N-terminus using the HotStar HiFidelity polymerase kit (Qiagen Inc., Hilden, Germany). Alanine (or glycine) substitutions in the mCCR8 N-terminal region were conducted using QuikChange Lightning Site-Directed Mutagenesis Kits (Agilent Technologies Inc., Santa Clara, CA, USA). PCR fragments bearing the desired mutations were inserted into the pCAG-Ble vector (FUJIFILM Wako Pure Chemical Corporation) using the In-Fusion HD Cloning Kit (Takara Bio, Inc., Shiga, Japan).
Antibodies
C8Mab-2 was established by the CBIS method. 23 NZ-1 (an anti-PA16 tag mAb) was described previously. 28 A secondary Alexa Fluor 488-conjugated anti-rat immunoglobulin G (IgG) was purchased from Cell Signaling Technology, Inc. (Danvers, MA).
Flow cytometry
Cells were harvested after brief exposure to 0.25% trypsin/1 mM ethylenediaminetetraacetic acid (Nacalai Tesque, Inc.). After washing with 0.1% bovine serum albumin in phosphate-buffered saline, cells were treated with C8Mab-2 (10 μg/mL) or NZ-1 (1 μg/mL) for 30 minutes at 4°C and subsequently with Alexa Fluor 488-conjugated anti-rat IgG (1:2000; Cell Signaling Technology, Inc.). Fluorescence data were obtained using the SA3800 Cell Analyzer (Sony Corp.).
Results
Determination of the epitope of an anti-mCCR8 mAb by flow cytometry using chimeric proteins
We employed the CBIS method to develop an anti-mCCR8 mAb (clone C8Mab-2). 23 C8Mab-2 is applicable for flow cytometry and immunocytochemistry. To investigate the binding epitope of C8Mab-2, we focused on four extracellular regions of mCCR8, including the N-terminal region [1–33 amino acids (aa)], extracellular loop 1 (ECL1; 92–105 aa), ECL2 (170–200 aa), and ECL3 (262–278 aa). The four extracellular regions of mCCR8 were substituted into the corresponding regions of mCCR3, which possesses a similar structure to mCCR8. As shown in Figure 1, mCCR3 (mCCR8p1–33), mCCR3 (mCCR8p92–105), mCCR3 (mCCR8p170–200), and mCCR3 (mCCR8p262–278) were generated. The chimeric proteins were transiently expressed on CHO-K1 cells, and the reactivities to C8Mab-2 were analyzed using flow cytometry (Fig. 2A). C8Mab-2 reacted with mCCR3 (mCCR8p1–33) and CHO/mCCR8 cells, but not with other chimeric proteins (Fig. 2A). The cell surface expression of each mutant was confirmed by an anti-PA16 tag mAb, NZ-1 (Fig. 2B). These results indicated that the N-terminal region of mCCR8 is recognized by C8Mab-2.
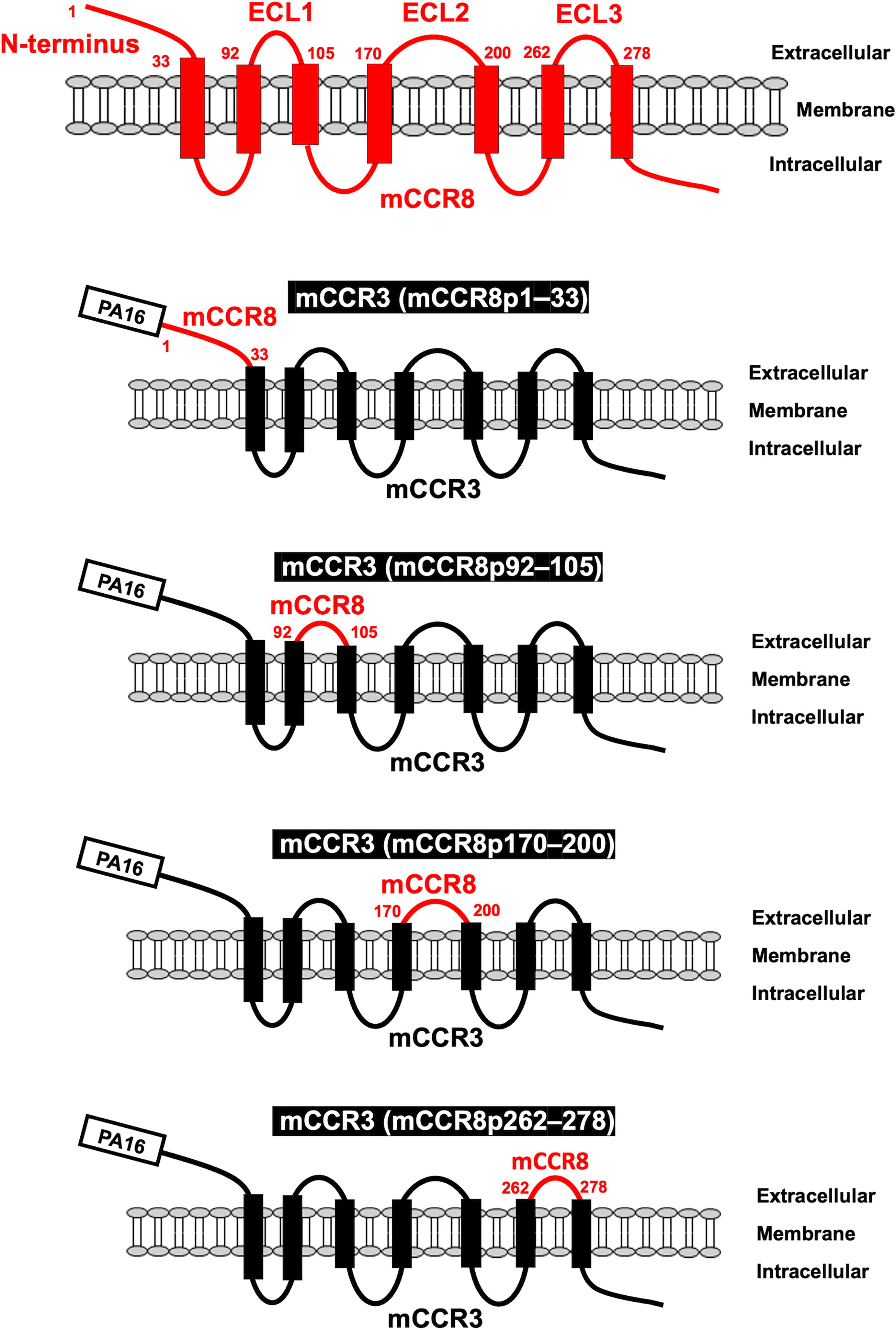
Schematic illustration of chimeric proteins. The four extracellular regions of mCCR8, including the N-terminal region (1–33 aa), ECL1 (92–105 aa), ECL2 (170–200 aa), and ECL3 (262–278 aa), were substituted into the corresponding regions of mCCR3. aa, amino acids; mCCR, mouse C-C motif chemokine receptor; ECL, extracellular loop.
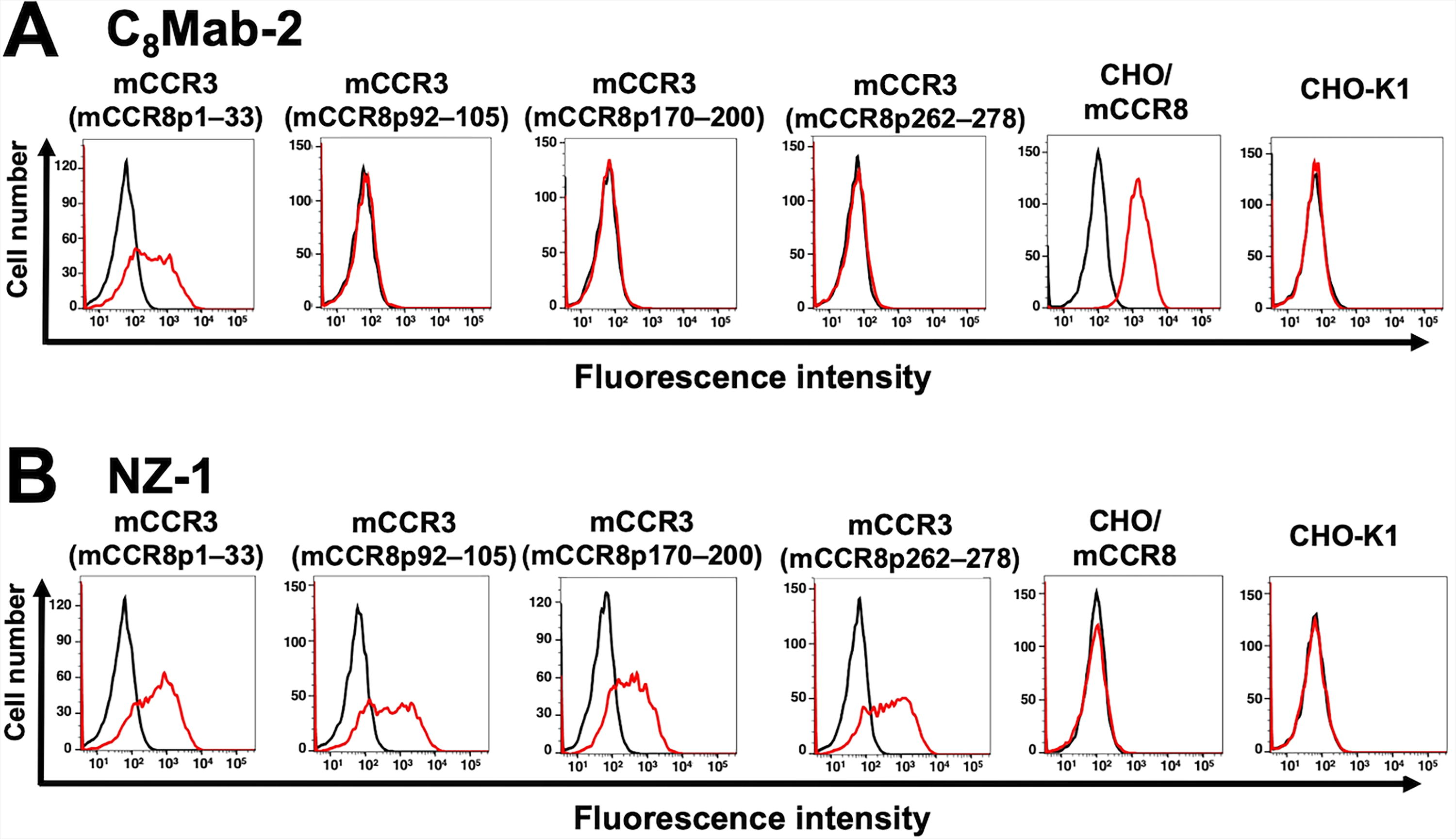
Determination of the epitope of an anti-mCCR8 mAb by flow cytometry using chimeric proteins. C8Mab-2 (10 μg/mL)
Determination of the C8Mab-2 epitope by flow cytometry using 1×alanine scanning
Next, 1×alanine scanning was conducted in the N-terminal region except for Cys23. Thirty-two 1×alanine (or glycine) substitution mutants of mCCR8 were constructed, and the mutant proteins were stably expressed on CHO-K1 cells. The reactivity against C8Mab-2 was assessed using flow cytometry. As shown in Figure 3A, C8Mab-2 did not react with a mutant (T20A). In contrast, C8Mab-2 reacted with the other 31 mutants. The cell surface expression of each mutant was confirmed by NZ-1 (Fig. 3B). These results showed that Thr20 of mCCR8 is important for C8Mab-2 binding.
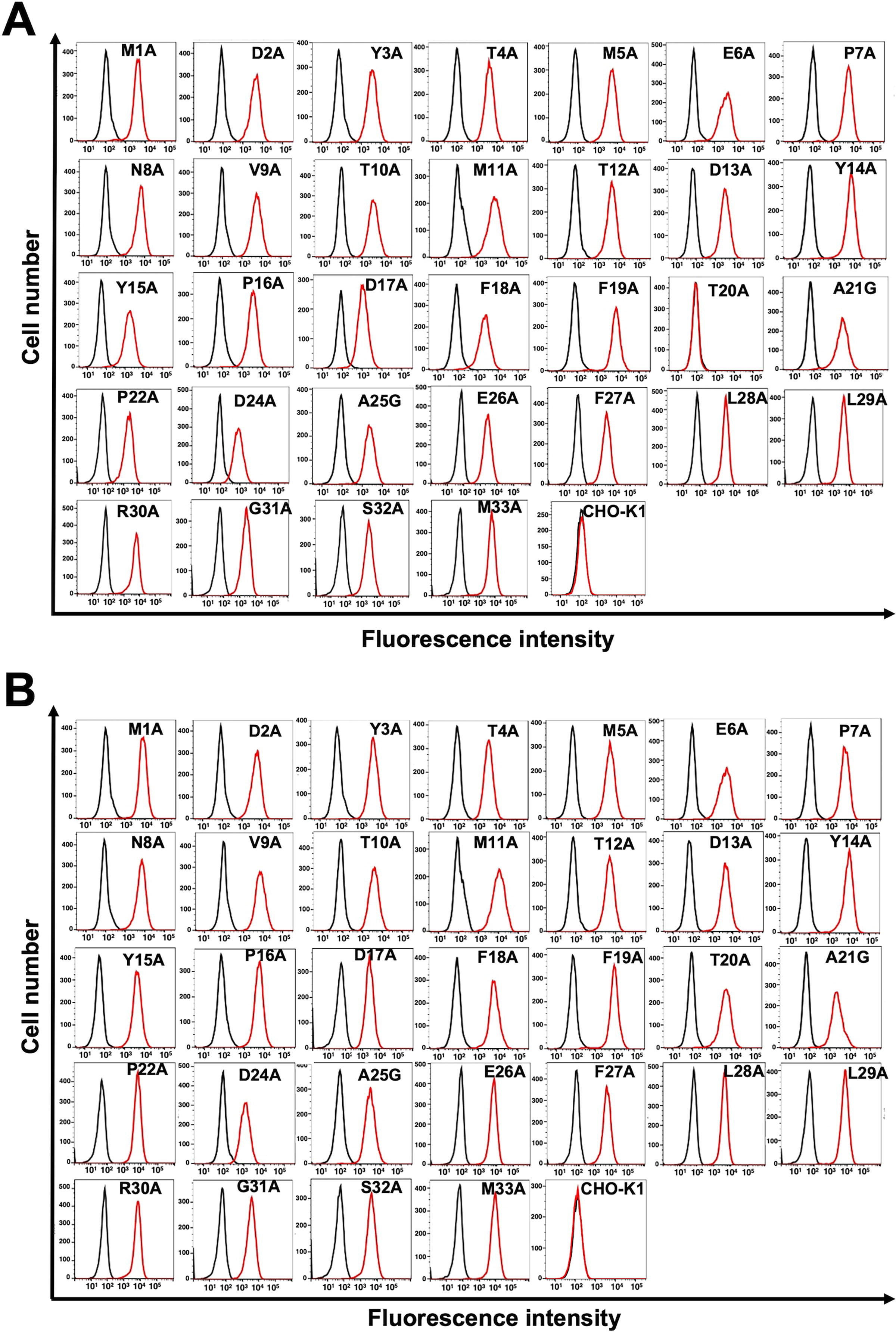
Determination of the C8Mab-2 epitope by flow cytometry using 1×alanine scanning. C8Mab-2 (10 μg/mL)
Determination of the C8Mab-2 epitope by flow cytometry using 2×alanine scanning
We also examined the reactivity of C8Mab-2 against 2×alanine (or glycine)-substituted mCCR8. We constructed thirty 2×alanine (or glycine)-substituted mutants in the N-terminal region of mCCR8 except for Cys23. The mutant proteins were transiently expressed on CHO-K1 cells. The reactivity against C8Mab-2 was assessed using flow cytometry. As shown in Figure 4A, C8Mab-2 did not react with the four mutants (D17A-F18A, F19A-T20A, T20A-A21G, and A21G-P22A). In contrast, C8Mab-2 reacted with the other 26 mutants. The cell surface expression of each mutant was confirmed by NZ-1 (Fig. 4B). These results showed that a motif from Asp17 to Pro22 in mCCR8 is important for C8Mab-2 recognition.
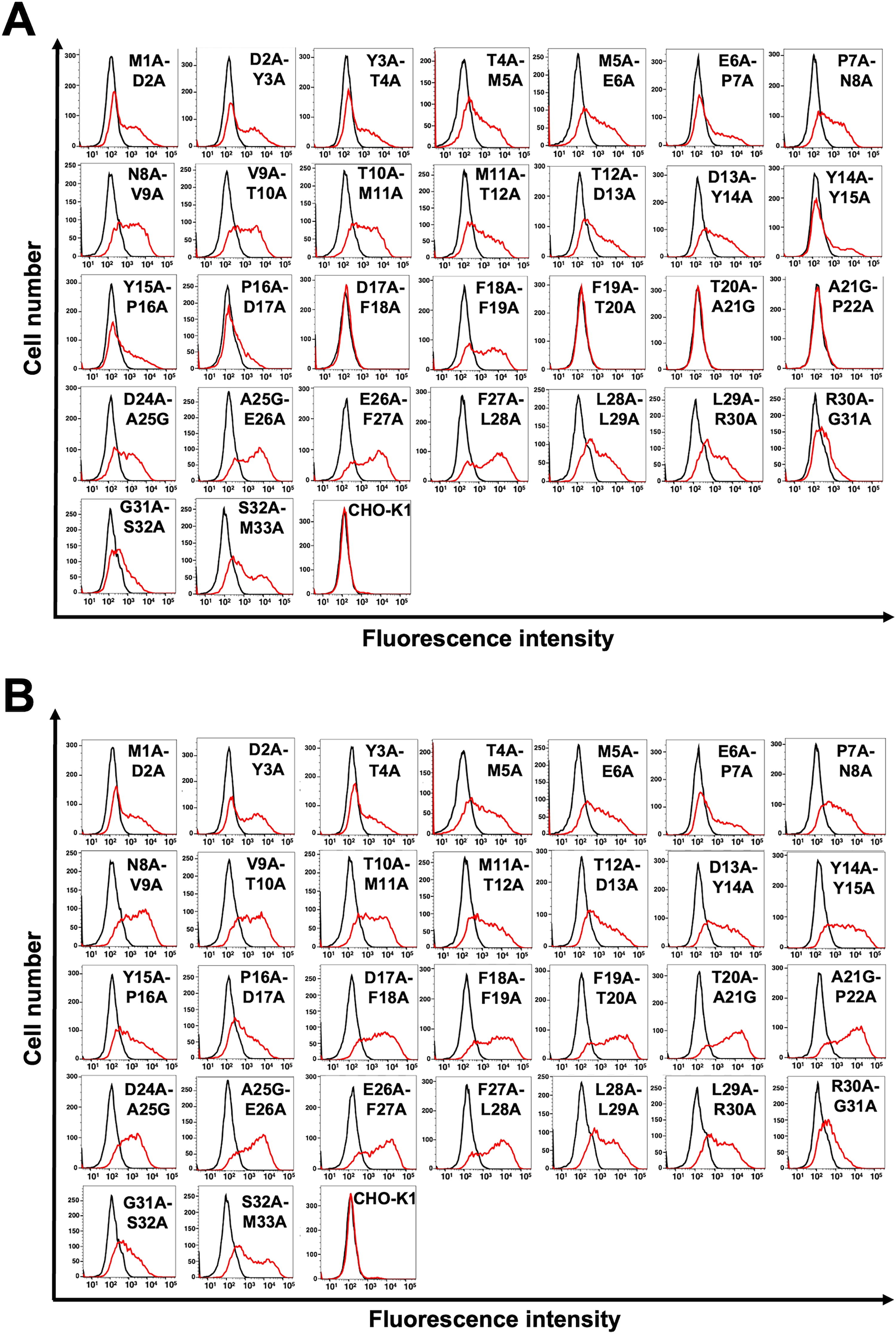
Determination of the C8Mab-2 epitope by flow cytometry using 2×alanine scanning. C8Mab-2 (10 μg/mL)
Discussion
In this study, we performed the flow cytometry-based epitope mapping of an anti-mouse CCR8 mAb (C8Mab-2) using the chimeric proteins (Figs. 1 and 2). Furthermore, we determined that the 17-DFFTAP-22 is important for the recognition by C8Mab-2 in the 2×alanine scanning (Fig. 4), and Thr20 is a central amino acid of the epitope by the 1×alanine scanning (Fig. 3). Figure 5 summarizes the epitope of C8Mab-2. In the epitope mapping of an anti-human CCR8 mAb (clone mAb1), CCR8 chimeras, in which the N-terminus, ECL1, ECL2, or ECL3 were replaced with the corresponding sequences from human CCR5, were used. 17 The mAb1 did not recognize ECL1-replaced CCR8 and ECL2-replaced CCR8, suggesting that both ECL1 and ECL2 are required for mAb1 binding. 17 Because we could not determine the binding epitope of our other anti-mCCR8 mAbs (C8Mab-1 and C8Mab-3) in this study (data not shown), the substitution of two ECLs may be required for the identification of the epitopes of those anti-mCCR8 mAbs.

Schematic illustration of the C8Mab-2 epitope, which was identified by 1×alanine scanning and 2×alanine scanning. 17-DFFTAP-22 is important for the recognition by C8Mab-2 in the 2×alanine scanning (underlined). Thr20 (red) is determined to be a central amino acid of the epitope by the 1×alanine scanning.
The “hot” tumor is characterized by the massive infiltration and enrichment with CD8+ effector T cells, which is important for the antitumor immune responses. Immune checkpoint inhibitors such as anti-PD-1 mAbs are effective in hot tumors. However, the response rate of immune checkpoint inhibitors is still low due to the lack of CD8+ effector T-cell infiltration or accumulation of Treg cells suppressing the effector activities, which is characterized as “cold tumors.”29,30 Since CCR8 expression is increased in tumor-infiltrated Treg cells, CCR8 is one of the promising target for depleting of Treg cells selectively in tumors. 4 Anti-mCCR8 mAbs have been used to suppress cancer growth in several cancer models.7,8,31 Furthermore, an anti-human CCR8 mAb (S-531011) was developed. 32 S-531011 has antibody-dependent cellular cytotoxicity activity against tumor-infiltrating CCR8+ Treg cells and neutralization activity against the CCR8 signaling. 32 Meanwhile, there is no information about the relationship between the Treg cells-depleting activity and epitope of the mAbs. Our strategy for epitope identification would contribute not only to the understanding of mAb-epitope interaction but also to the improvement of those therapeutic mAbs.
Footnotes
Author’s Contribution
H.K. and T.T. performed the experiments. M.K.K. and Y.K. designed the experiments. H.S. and M.K.K. analyzed the data. H.S. and Y.K. wrote the article. All authors have read and agreed to the published version of the article.
Author Disclosure Statement
The authors have no conflict of interest.
Funding Information
This research was supported in part by