
Abstract
Mammalian cell line stability is an important consideration when establishing a biologics manufacturing process in the biopharmaceutical and in vitro diagnostics (IVD) industries. Traditional Chinese hamster ovary (CHO) cell line development methods use a random integration approach that requires transfection, selection, optional amplification, screenings, and single-cell cloning to select clones with acceptable productivity, product quality, and genetic stability. Site-specific integration reduces these disadvantages, and new technologies have been developed to mitigate risks associated with genetic instability. In this study, we applied the Leap-In® transposase-mediated expression system from ATUM to generate stable CHOK1 pools for the production of four recombinant antibody reagents for IVD immunoassays. CHO cell line stability is defined by consistent antibody production over time. Three of the CHOK1 pools maintained productivity suitable for manufacturing, with high antibody yields. The productivity of the remaining CHOK1 pool decreased over time; however, derivative clones showed acceptable stability.
Introduction
A clonally derived stable cell line is defined as a homogeneous cell population that retains 70% or more of its original volumetric productivity titer over 70 generations. 1 Establishing a stable cell line is a critical step in the manufacture of biological therapeutics in the biopharmaceutical industry or biological reagents for in vitro diagnostics (IVD) use. While E. coli and yeast expression systems are cheaper and faster for large-scale protein expression, the manufacture of multisubunit biological products, such as antibodies requires fine control of gene expression ratios, making mammalian cells better hosts. The Chinese hamster ovary (CHO) cell remains a predominant system in the biomanufacturing field,2,3 and several stable CHO cell systems are available that work well in industry, such as the CHO-GS and CHO-DHFR systems.4,5 For therapeutic drug manufacturing, a stable CHO cell line originating from a single cell (clone) is required to minimize product heterogeneity and reduce safety and immunogenicity risks. In the IVD field, specific biologics produced by stable CHO cell lines include antibodies and antigens for immunoassays, and antibodies for immunohistochemistry.
Recombinant CHO stable cell lines that produce specific biological products are primarily generated by random target gene integration. However, this approach is time-consuming, and involves transfection, selection, cloning, and identification of clones with the required product titer, quality, and stability. To bring innovative biologics and health care products to patients more quickly, for example, in the development of diagnostic assays during the SARS-CoV-2 pandemic, new targeted approaches were needed to rapidly create stable cell lines for biological manufacturing. Transposase-mediated semi-targeted transgene integration has been shown to generate stable CHO cell lines faster than random gene targeting, achieving high production rates at a lower cost.6,7 ATUM (Newark, CA) developed the Leap-In® expression system based on transposase technology to create stable CHO cells with relatively high and uniform integration sites, resulting in high production yields even at the stable pool stage. We asked whether production of antibodies by stable CHO pools, rather than traditional clonal CHO cell lines, could be applied to the manufacture of IVD assay reagents. We utilized the Leap-In system to generate four different recombinant antibody-producing CHOK1 cells and compared stable pool and single cell clone stability and antibody product parameters.
Materials and Methods
DNA construction and cloning
All DNA sequences were chemically synthesized from phosphoramidites, cloned into intermediate vectors, and transformed into E. coli hosts using ATUM’s proprietary technology. To establish a stable pool, a CMV plasmid construct based on the pD2596ht+f-CMV(Mm) vector configuration was designed to express glutamine synthetase for each of four monoclonal antibodies: anti-SARS-CoV-2 nucleocapsid human IgG1λ, 8 anti-T4 sheep IgG1λ, anti-human IgG4 mouse IgG1k, and anti-BNP mouse IgG1k. Coding sequences were assembled by overlapping oligonucleotides and PCR, which were then cloned into intermediate vectors. From the intermediate vectors, the inserts were transferred to a final expression construct. Molecular cloning was performed following the Electra cloning system using Type IIS restriction enzymes (ATUM). Taking the anti-BNP mAb as an example, genes encoding anti-BNP heavy chain (HC) and light chain (LC) were cloned into vector pD2596ht+f-CMV(Mm) to generate the construct. The DNA sequences of the HC and LC in the expression plasmid were confirmed by Sanger sequencing and were found to be 100% identical to the designed sequence. A large-scale plasmid prep was performed to generate stable pools. The DNA sequence of the entire stable expression plasmid was confirmed by nanopore sequencing using the Oxford Nanopore Technologies platform (Oxford, UK).
Stable CHO Pool development
CHOK1 glutamine synthetase knockdown (miCHO-GS) cells were maintained in shake flasks in growth medium: Ex-CELL Advanced Fed-batch Medium (Millipore Sigma, Burlington, MA) with 4 mM glutamine. Cells were grown on a Monday–Friday passage schedule and kept in shaker incubators at 37°C and 5% CO2, with 70–80% relative humidity. Transfections were performed by electroporation. After a recovery period, the transfectant cultures were introduced to metabolic selection by media exchange to a glutamine-free formulation. Selection medium was replaced every 5 days until the cultures recovered. Throughout this process, the cultures were maintained at 37°C and 5% CO2, with 70–80% relative humidity. Upon reaching ≥80% viability, the recovered stable pools were transferred to 125-ml shaker flasks. CHOK1 cells from the stable pools were cryopreserved.
Cell culture and cell passage study
CHOK1 stable pools were quick thawed in a 37°C water bath, then centrifuged at 1000 rpm for 5 minutes. The cell pellet was resuspended in growth medium and maintained at 37°C and 5% CO2, with 70–80% relative humidity in 125-ml shaker flasks at 150 rpm. The pools were passaged at least twice before seeding at 0.5x106/mL for the cell passage study. The cells were passaged every 2–3 days, seeded at 0.5x106/mL, then allowed to grow for 7 days (Fig. 1A). At each passage, cells were transferred to another flask at 0.5x106/mL to serve as the parental flask. At the end of each 7-day culture, the cells were centrifuged at 1000 rpm for 5 minutes and the supernatant was collected for quantitation by size-exclusion high-performance liquid chromatography (SEC-HPLC)-IgG.

Cell passage study.
In our study, we performed a total of 20 cell passages for each cell pool. The experiment was specifically designed to cover the entire lifespan of CHOK1 cells, which are commonly used in standard IVD industry practices. Typically, the production volume for a specific diagnostic immunoassay involves antibody production at a scale of under 100 liters. During the scaling process, it takes approximately 10 passages to transition from a discovery cell bank to a production cell bank. Subsequently, an additional 10 passages (or fewer) are needed to further scale up the production cell bank to achieve a final CHOK1 cell volume ranging from 10 to 100 liters. By conducting 20 passages instead of the conventional 30, we aimed to assess cell stability. Specifically, a 30% reduction in recombinant antibody yield from passage 1 to passage 20 would indicate that the CHOK1 cell pool is not suitable for manufacturing purposes.
HPLC-IgG quantitation
All antibody quantitation was performed on a Waters HPLC (Milford, MA), with an analytical Protein G Column (Thermo Fisher, Waltham, MA). Briefly, 200 µL of the collected CHOK1 cell culture supernatant was loaded onto the column and eluted with 12 mM HCl solution. A mouse IgG antibody mixture was used as a calibrator, diluted from 12.5 µg/mL to 400 µg/mL.
Anti-BNP antibody purification by protein a
CHOK1 cell culture supernatant was centrifuged and filtered. The supernatant was doubled with a binding buffer. The prepared supernatant was loaded onto a pre-equilibrated 5-ml Protein A column using the ÄKTATM automated purification system (GE Healthcare, Pittsburgh, PA). The column was then washed with 5–10 volumes of the binding buffer and the antibody was eluted with IgG elution buffer. The antibody was further dialyzed against PBS using a 10 KDa molecular weight cutoff membrane.
Ion exchange chromatography (IEx)
Protein A column purified anti-BNP antibody was loaded at 5 mL/min onto a GE HiTrap SP HP (5 mL) column previously equilibrated with binding buffer 20 mM sodium acetate (NaOAc) using the AKTA Prime Plus (Cytiva, Marlborough, MA). After washing with 40 mL of binding buffer, gradient elution was performed with a buffer (20 mM NaOAc/1M NaCl) in which NaCl was increased from 0% to 20% over the first 50 mL, then to 40% over the final 250 mL. Fractions were collected and analyzed for the presence of antibody.
Capillary electrophoresis sodium dodecyl sulfate (CE-SDS) analysis of purified anti-BNP antibody
All experiments were performed on the PA 800 plus Pharmaceutical Analysis System (SCIEX, Framingham, MA). Bare, fused-silica capillaries of 50 μm ID × 20 cm to detection, were used for the separation. To prepare the SDS–protein complex, the protein sample was desalted and concentrated using a 3k-cutoff filter membrane to 5 mg/mL in 100 mM Tris-HCl pH 9.0, 1% SDS. The final concentration of the sample in the sample injection matrix was 1.0 mg/mL. The IgG–SDS complex was reduced by adding 5% neat 2-mercaptoethanol (v/v) then heating in a boiling water bath for 3 minutes. For a nonreduced sample, the IgG–SDS complex was first alkylated with iodoacetamide and then heated at 70°C for 3 minutes.
An optimized separation method and sequence were set up for batch analysis of 24 samples at a time. For each separation cycle, the capillary was first preconditioned with 0.1N NaOH, 0.1N HCl, deionized water, and SDS gel buffer. All gel buffers were degassed for 2 minutes under vacuum before use. Electrophoresis was performed at a constant −15 kV with the capillary kept at 25°C using recirculating liquid coolant. Signal generation through a photodiode array at 220 nm and 2 Hz was used for electropherogram visualization.
Capillary isoelectric focusing (cIEF) analysis of purified anti-BNP antibody
The cIEF was performed using the Maurice instrument (Bio-Techne, Minneapolis, MN) according to the manufacturer’s instructions. Briefly, antibody samples diluted to 0.25 mg/mL in CE-grade water were loaded onto sample vials with pI standards, appropriate ranged ampholytes, and 1% methyl cellulose. Samples were run and analyzed using Compass Software (Bio-Techne). Samples were focused for 1 minute at 1500V and then 10 minutes at 3000V. Native fluorescent detection was employed using a 30-s exposure time to the CCD camera.
Size-exclusion high-performance liquid chromatography (SEC-HPLC) of the anti-BNP antibody
SEC-HPLC was performed on a Waters HPLC system Model e2695 equipped with a Waters PDA 2998 (Milford, MA). Samples were diluted to 1 mg/mL in HPLC-grade water. Samples were injected onto a TSKgel G3000 SWXL column (Tosoh Bioscience, South San Francisco, CA) with 150 mM NaCl, 100 mM phosphate pH 7.2 at a flow rate of 1 mL/minute at room temperature. All chromatograms were generated using Empower 3 software (Waters, v3.6.0.)
Electrospray ionization mass spectrometry (ESI-MS)
Purified anti-BNP monoclonal antibody samples were desalted using a 10k-cutoff filter before injection. All experiments were performed on an AB SCIEX Triple TOF 5600 instrument equipped with an Eksigent microLC 200 (SCIEX). Samples were analyzed intact as well as after reduction with TCEP with a 5-µL injection. Solvent A was 0.1% formic acid in 95% water and 5% acetonitrile, and Solvent B was 0.1% formic acid in 95% acetonitrile and 5% water. All samples were subjected to the gradient before detection.
Anti-BNP-mIgG1k binding kinetics study
Surface plasmon resonance measurements were carried out on a BIAcore T200 automated system (BIAcore, Cytiva Life Sciences, Uppsala, Sweden) using anti-mouse IgG-coupled CM5 Series S sensor surfaces. The anti-mouse sensor surfaces were prepared according to the manufacturer’s instructions using Mouse Antibody Capture and Amine Coupling Kits, both obtained from BIAcore. Anti-BNP antibody-binding interactions were conducted in HBS-EP+ buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% P20, pH 7.4,) supplemented with 0.1% BSA and 0.1% carboxymethyl dextran and analyzed using BIAcore T200 evaluation software (v3.2.1).
Anti-mouse IgG-coupled sensor surfaces (FC1 and FC2) were prepared by activating a CM5 Series S sensor chip with aqueous 0.2M EDAC/0.05M NHS solution for 7 minutes at 10 µL/min. Polyclonal anti-mouse IgG (30 µg/mL in 10 mM NaOAc, pH 5.0) was subsequently injected over the sensor surfaces for 7 minutes at 10 µL/min followed by capping of nonreacted sites with 1M ethanolamine, pH 8.5 for 7 minutes. The coupling resulted in immobilization of 8190 response units (RU) of anti-mouse IgG coupled to FC1 and 6621 RU to FC2 of the sensor surfaces.
All anti-BNP antibody-binding evaluations were conducted at 25°C using the anti-mouse IgG capture surfaces prepared above. Initially, a nonspecific mAb (20 µg/mL in running buffer) was captured on FC1 at 10 µL/min for 300 seconds followed by a 300-s stabilization period. Anti-BNP antibody (25 µg/mL) was subsequently captured on FC2 at 10 µL/min for 300 seconds followed by a 300-s stabilization period, which provided approximately equal captured antibody responses on the two flow cells. Human BNP-32 (Peptide Institute, Inc., Osaka, Japan) was then injected across both surfaces at 100 µL/min for 120 seconds with a 300-s dissociation period. Seven concentrations of human BNP-32 (300–0.411 nM, 3-fold dilutions) were randomly injected in duplicate for each anti-BNP antibody series being evaluated. Zero-antigen controls were also run at the beginning and end of each series. Sensor surfaces were regenerated after each binding evaluation by injecting 10 mM glycine, pH 1.7 at 20 µL/min for 3 minutes followed by a 1-min stabilization period. All binding evaluations were double reference-corrected using data from FC2-1 and fit to a 1:1 binding model to determine association and dissociation rate constants (ka and kd) and the binding affinity (KD).
Results
CHOK1 pools with stable productivity
Three of the recombinant antibody CHOK1 pools showed acceptable stability after 20 cell passages (Fig. 1B). The average production of anti-SARS-CoV-2 nucleocapsid human IgG1λ by the CHOK1 pool was 755 mg/L, from 742 mg/L at the beginning to 749 mg/L at the end of 20 passages. The difference in production between passage 1 and passage 20 was 0.9%. Production of the anti-T4 sheep IgG1λ/CHOK1 pool decreased by approximately 22.7% during the 20 passages, from 727 mg/L to 562 mg/L, with an average production of 594 mg/L. Production of the anti-human IgG4 mouse IgG1k/CHOK1 pool decreased slightly (6.6%) during the study, from 527 mg/L to 492 mg/L, with an average production of 533 mg/L.
CHOK1 Pool with decreasing productivity and the effect of glutamine on productivity
The anti-BNP mouse IgG1k antibody CHOK1 pool was not stable in the presence or absence of
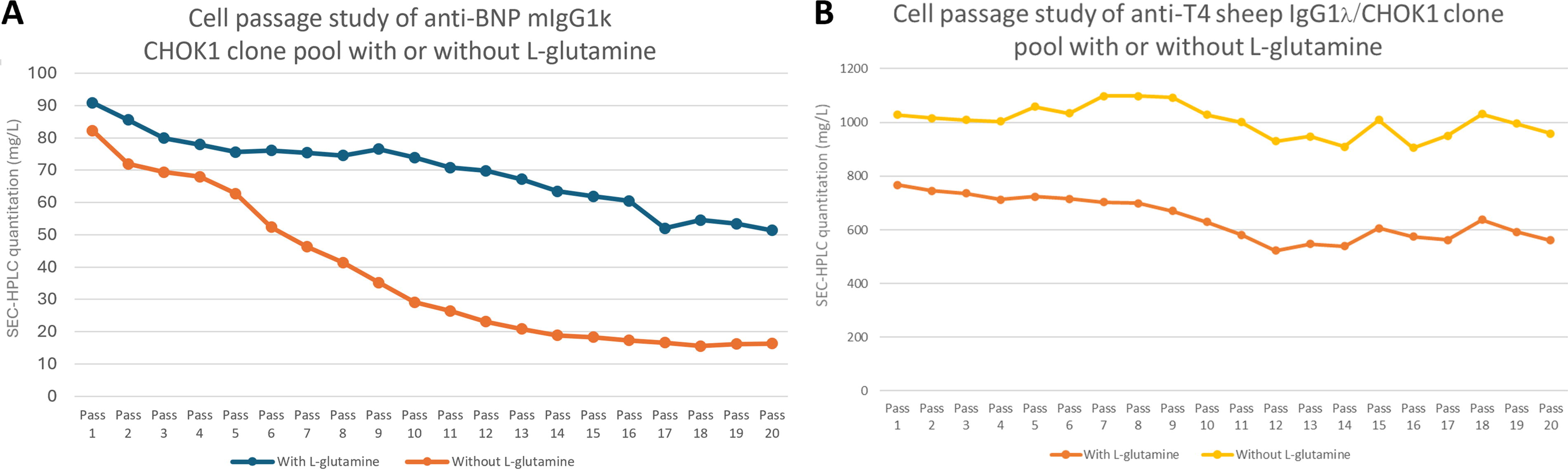
CHOK1 pool stability in the presence or absence of
While the anti-BNP mouse IgG1k CHOK1 pool showed less stability in the absence of
Anti-BNP mIgG1k CHOK1 clone line stability
Based on the cell passage study results that showed instability of the anti-BNP mIgG1k CHOK1 pool, we cloned the pool using ClonePix FL. After 7 days of growth on soft agar plates, some single-cell colonies were observed with strong fluorescence halos, indicating high production (Fig. 3A). Approximately 300 clones were picked by ClonePix based on colony fluorescence intensity. Three clones (r259, r288, and r387) were selected by enzyme immunoassay during the clone expansion and studied in culture medium without

Stability of anti-BNP mIgG1k/CHOK1 clones.
Anti-BNP mIgG1k antibody-binding kinetics
We evaluated the binding kinetics of the anti-BNP mIgG1k antibody produced by the CHOK1 pool and the CHOK1 clone r288. Binding to human BNP-32 antigen was evaluated using an automated BIAcore T200 system with anti-mouse IgG capture sensor surfaces. Anti-BNP antibody from the CHOK1 pool as well as the CHOK1 clone had equivalent ka, kd, and KD values (Fig. 4).

Anti-BNP mIgG1k antibody-binding kinetics.
The anti-BNP mIgG1k antibodies produced by the CHOK1 pool and clone were further characterized as shown in Table 1 and Figure 4. Analytical characterization using various methods showed that the antibodies from each source had very close similarity.
Characterization of Anti-BNP mIgG1k Antibody Products
CE-SDS, capillary electrophoresis sodium dodecyl sulfate; cIEF, capillary isoelectric focusing; LC, light chain; HC, heavy chain.
Discussion
In this study, we utilized the Leap-In® transposase-mediated stable integration system to establish stable CHOK1 pools that produce four different recombinant antibody products. The productivity of three of the four CHOK1 pools was stable for 20 cell passages over approximately 50 days (50 doubling times or generations). One CHOK1 pool was unstable, with decreasing anti-BNP mouse IgG1k productivity, and was therefore cloned. The productivity of the selected derivative clones was found to be stable for 20 passages. The anti-BNP mouse IgG1k/CHOK1 pool and CHOK1 clone produced anti-BNP antibody products with identical binding characteristics.
Currently, clonally derived stable cell lines are the most common cell substrates used for biopharmaceutical protein manufacturing. Clonality is very important for cell-based production of therapeutic protein products, as clonal lineages ensure consistent product quality and thus product efficacy and safety. However, establishing clonal cell lines is time- and resource-consuming, so efforts have focused on the utility of stable pools as cell substrates. These efforts were intensified during the COVID-19 pandemic because of the need for extremely rapid development and production of antibodies for therapeutic and diagnostic purposes. 9 The Leap-In system generates stable pools of individual clones that have much more homogeneous distributions of productivity and product quality than those derived from conventional random integration-based methods. 10 The homogeneity of clones in these stable pools means that the pools and the clones derived from them have remarkably comparable productivity and product quality, making them excellent candidates for replacing clonal cell lines for cell-based production of therapeutic and diagnostic protein products.
The cell passage study is a conventional method to evaluate mammalian cell line stability, an important feature for cell lines used in biomanufacturing. A stable cell line can be defined as a clonally derived homogeneous cell population that retains at least 70% of its volumetric productivity titer over 70 generations. Typically, 60 generations are required to scale up from a master cell bank to production volume and demonstrate no clinically meaningful differences from reference product as estimated by structure, function, purity, chemical identity, and bioactivity. 1 In the IVD industry, the mammalian cell line-based manufacturing scale is typically lower than that in the biopharmaceutical industry, ranging from 3L to 100L. Most IVD cell culture scales are under 10L to go from master cell bank to production volume within 50 days. Therefore, 20 cell passages (∼50 generations) is sufficient for our CHO cell line stability test.
What factors affect cell line production stability? In the case of clonally derived cell substrates, production stability depends upon the clone’s genetic stability, which in turn is influenced by the structure of the integrated transgene (concatemeric repeats tend to recombine out over time and become unstable), epigenetic silencing, chromosome loss, and other genetic factors. The production stability of stable pools depends not only on the stability of the individual clones of the pool, but also on the population dynamics of those clones. Since ∼97% of the Leap-In mediated stable clones are genetically stable (there are no concatemers, and silencing is prevented by a combination of the Leap-In® transposase’s preference for integrating transposons into open chromatin and anti-silencing elements within the transposons), the stability of pool productivity is determined primarily by population dynamics. In general, clones with lower productivity grow faster than high producer clones because fewer resources are devoted to expressing the integrated transgene. Thus, even in a relatively homogeneous stable pool, the lower producers will eventually take over the population resulting in lower pool productivity.
Three of the four antibody-producing Leap-In® generated pools in this study—showed highly stable productivity, with the anti-SARS-CoV-2, the anti-T4, and the anti-human IgG4 antibody pools losing only 0.9%, 22.7%, and 6.6% of starting activity, respectively, during 20 passages. In contrast, the pool expressing the anti-BNP mouse IgG1k was highly unstable, losing 50–80% of productivity in the same time period. This appears to be because producing high levels of the anti-BNP mouse IgG1k is toxic to the CHOK1 cells. After transfection, the pool expressing the anti-BNP mouse IgG1k took nearly twice as long as the other three pools to recover with no glutamine selection (25 days for anti-BNP compared with 15 days for the others, data not shown), and the initial titer from the pool was approximately 10% of the titers for the other three antibodies. These are hallmarks of cell pools that are trying to make proteins whose expression is toxic to the host cells, for example, because of induction of the unfolded protein response. The longer recovery period appears to be the time it takes to select cells that express enough glutamine synthetase to provide the glutamine the cells need to grow, but low enough expression of the transgene to avoid cell death from protein-associated toxicity. Such difficult-to-express proteins will obviously amplify population dynamic changes. Since there is a high fitness penalty for expressing the anti-BNP antibody, cells that express very little of it will be at a strong selective advantage and will overgrow the population quickly, resulting in high instability of pool productivity. This probably also explains the observation that this pool was even more unstable in the absence of glutamine; lack of exogenous glutamine probably caused additional stress to cells and made it more difficult for producing cells to compete with the nonproducing population. In our study, we utilized the anti-T4 sheep IgG1 CHOK1 pool as a control experiment to investigate the impact of glutamine. Surprisingly, the data revealed no stability impact in this context. However, it is important to note that we did not assess the effect of glutamine on the anti-SARS-CoV-2 and antihuman IgG4 antibody pools. These pools were rapidly recovered from selection in a medium without glutamine.
The Leap-In® transposase-mediated stable integration system, which is considered semi-targeted integration, demonstrated reduced phenotypic heterogeneity compared with random integration. Notably, the recombinant antibodies produced from stable pools exhibited comparable product quality attributes when compared with clonal cell lines. 7 Despite the instability of the anti-BNP pool, clones derived from the pool were stable. This is because clonal instability requires genetic mechanisms for reducing transgene expression. As described above, Leap-In® transposition avoids the root causes of genetic instability, even in cases where production of a protein is disadvantageous to a cell. Finally, as another example of pool-to-clone comparability, the binding characteristics of the pool derived and the clonal anti-BNP proteins were identical.
In conclusion, using transposon-based systems, recombinant stable CHOK1 pools could be generated in 2–3 weeks, saving time over conventional methods while achieving very high product titers. CHOK1 pools that are stable for 20 passages are therefore a viable option for rapid recombinant protein production in IVD biologics manufacturing.11,12
Footnotes
Acknowledgment
The authors acknowledge Stacey Tobin, PhD, for scientific writing assistance in the preparation of this article.
Authors’ Contributions
B.T.: conducted the study, analyzed the results, and wrote the article (lead); Z.L.: conducted the study, analyzed the results, and reviewed the article (equal); J. Moore: conducted the study, analyzed the results, and reviewed the article (equal); A.K.S.: conducted the study and analyzed the results (equal); M.J.W.: conducted the study and analyzed the results (equal); D.Y.M.: conducted the study and analyzed the results (equal); M.G.: conducted the studyand analyzed the results (equal); W-C.L.: conducted the study and analyzed the results (equal); J.B.: conducted the study and analyzed the results (equal); T.S.: conducted the study and analyzed the results (equal); Y.N.C.P-G.: conducted the study and analyzed the results (equal); R.B.: conducted the study and analyzed the results (equal); T.R.: conducted the study and analyzed the results (equal); S.M.: analyzed the results and reviewed the article (equal); P.H.: analyzed the results and reviewed the article (equal). All authors approved the final version of the article for submission.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author Disclosure Statement
B.T., Z.L., J. Moore, A.K.S., M.J.W., D.Y.M., M.G., W-C.L., J.B., T.S., Y.N.C.P-G., R.B., T.R., S.M., and P.H. are employees of Abbott Laboratories and J. Minshull, F.B., and V.S. are employees of ATUM.
Funding Information
The study was funded by Abbott Laboratories.