
Abstract
Emergence of antimicrobial resistance is among the most worrisome issues in public health worldwide. Vancomycin resistance is rapidly spreading, resulting in increased morbidity, mortality, and healthcare-associated costs. Multiple strategies are required to preserve the effectiveness of this essential antibiotic. It has been recently shown that biliary excretion of vancomycin following parenteral administration results in significant fecal concentrations of vancomycin that may lead to selection of vancomycin-resistant strains within the colon. In this study we present a novel strategy for preventing this undesired effect and its consequences, using chemical trapping of vancomycin by a tripeptide analog that mimics the natural bacterial vancomycin binding-site. Initially, we demonstrated that a tripeptide analog can neutralize vancomycin activity against Enterococci at a molar excess of 28. In the second phase, two chemical modifications, designed to attach the tripeptide to vancomycin covalently, were explored. Attachment of a 4-flurosulfonyl-benzoic acid (FSBA) moiety to the parent tripeptide resulted in vancomycin neutralization at a molar ratio of less than 4:1. Finally it was shown that the FSBA-bound tripeptide analog can prevent in-vitro selection of vancomycin-resistant Enterococci (VRE) from a mixed vancomycin susceptible/resistant population following exposure to vancomycin. These findings demonstrate the ability of the proposed strategy to prevent selection of VRE. The present proof-of-concept study provides the basis for further development of the proposed strategy. Further, this strategy may be implemented for combating resistance to other antimicrobials.
Introduction
De-novo occurrence of vancomycin resistance is considered a rare, complex phenomenon. Hence, it is currently assumed that the widespread presence of vancomycin-resistant bacteria is fundamentally the consequence of transmission and selection of resistant bacteria originating from a limited number of de-novo resistance mutations. 6
The colon hosts the largest physiological reservoir of bacteria within the human body. Enterococci are an abundant component of the human colonic microflora, generally exhibiting susceptibility to vancomycin. However, emergence of colonic VRE is a well-known consequence of several influences, such as exposure to glycopeptides and to other antimicrobial classes.12,14 It is widely accepted that rectal colonization consists a major reservoir of VRE within healthcare institutions. As the vast majority of vancomycin clinical use is intravenous (IV), the emergence of VRE following this route of administration requires sufficient biliary excretion of vancomycin, to provide the exposure necessary to promote selection of VRE. Indeed, a recent study has shown that IV vancomycin administration to patients results in considerable fecal vancomycin concentrations (3.3–94.4 mcg/ml following ≥5 days of 1 g twice-daily [BID]). 9 The presence of clinically-significant amounts of vancomycin within the colon following IV administration, coupled with the high-volume use of vancomycin, may facilitate the selection and emergence of vancomycin resistance.
A variety of strategies have been suggested for combating the global spread of antimicrobial resistance, among which is administration of higher antimicrobial doses. For vancomycin, recent evidence has shown that increased dosing is associated with a higher incidence of nephrotoxicity 21 and ototoxicity. 13 To preserve vancomycin as a useful tool in the ongoing struggle against pathogenic bacteria, additional tactics need to be developed and employed.
The colonic microflora is known to possess the inherent capability of resisting colonization of “nonnative” bacteria, a phenomenon termed “colonization resistance.” 10 However, this naturally-occurring reaction to invasion of extrinsic bacteria might be disturbed by antimicrobials reaching the colon, due to their effect on the natural microflora. This may result in long-term ecological consequences including establishment of colonization of resistant strains of potentially-pathogenic bacteria.
For prevention of this undesired consequence of antimicrobial administration, a novel approach has been recently proposed in relation to beta-lactam antimicrobials. This strategy is based on the concomitant administration of beta-lactamase as a gastrointestinal degrading system for those portions of beta-lactams reaching the colon following IV administration 16 or, as demonstrated by previous work of our laboratory, for the unabsorbed portion of an orally-administered beta-lactam. 17 These studies have shown that although the beta-lactam alone disrupts the colonic microflora, concomitant administration of beta-lactamase to the colon preserves its baseline bacterial composition and maintains colonization resistance. The implications of this strategy have been recently shown to extend beyond that of reducing resistance to beta-lactams and also into prevention of colonization of C. difficile. 25
In the present study we sought to employ a principally similar approach for trapping vancomycin within the gastrointestinal tract. We hypothesized that inactivation of vancomycin within the gastrointestinal tract may minimize VRE selection under the selective pressure induced by the undesired exposure to gastrointestinal vancomycin.
The objective of the present study was to design and test a chemical platform for vancomycin trapping. As vancomycin is not known to be enzymatically degraded by bacteria, a different, nonenzymatic approach is suggested: design of a compound mimicking the naturally-occurring binding site of vancomycin within the bacterial cell wall. Vancomycin inhibits bacterial wall synthesis by binding to bacterial acetyl-

Structures of the chemical components used in this study, as detailed in Table 1. FSBA, -4-(fluorosulfonyl)-benzoic acid.
a, D-Ala; Ac, acetyl; Br-Ac, bromoacetyl; FSBA, 4-(fluorosulfonyl)benzoic acid.
The present study was designed to address the following issues:
1. Can the extrinsic peptide Nα,Nε-diacetylated-K-a-a (diacetylated tripeptide, Peptide-1, Table 1) antagonize vancomycin's in-vitro antibacterial effect on Enterococcus faecalis, and, if so, in what molar ratio? 2. Can either acetyl group of the tripeptide (or both) be replaced by an active moiety, allowing for enhanced binding affinity to vancomycin and antagonize its' antibacterial effect in-vitro against E. faecalis and Enterococcus faecium in a lower molar excess than that of Peptide-1 alone? 3. Can the tripeptide prevent in-vitro selection of VRE following exposure of mixed susceptible/resistant Enterococci populations to vancomycin?
Materials and Methods
Chemistry
All starting chemicals were purchased from commercial sources and were used without further purification. 4-(fluorosulfonyl)benzoic acid (FSBA) was purchased from Sigma-Aldrich.
Peptide-1 (tripeptide Nα,Nε-diacetyl-K-a-a, see Table 1 for abbreviations) and its derivatives were synthesized on Trityl Resin using standard Solid Phase Peptide Synthesis procedures with Fmoc chemistry. 5 Mass spectra were recorded on a PerSeptive Biosystems MALDI-TOF MS, using α-cyano-4-hydroxycinnamic acid as matrix. Peptides were purified by preparative high-performance liquid chromatography.
Antagonizing of vancomycin antibacterial activity versus E. faecalis by Peptide-1
For assessment of vancomycin deactivation by Peptide-1, a standard inoculum of 5×105 colony-forming units (CFU) of E. faecalis ATCC 29212 was spread onto standard Mueller-Hinton (MH) agar plates (Novamed). Following overnight incubation at 35°C, wells of a volume of approximately 30mcl were created in the agar using sterile glass pipettes.
To use vancomycin in an amount equivalent to that of 30 mcg used in standard disk-diffusion clinical microbiology assays, a 10 mcg/mcl vancomycin solution (Vanco-Teva; Teva Pharmaceuticals) was prepared and 3 mcl aliquots (containing 30 mcg) were added to a series of Peptide-1 solutions to form 25 mcl aliquots containing Peptide-1 in the following vancomycin/Peptide-1 molar ratios: 2:1, 1:1, 1:5, 1:10, 1:28, and 1:56. Two such sets were prepared; one for immediate use and the second for preincubation at 35°C for 1 hour, to further facilitate the reaction between vancomycin and Peptide-1. Subsequently, the 25 mcl vancomycin-tripeptide aliquots were transferred to the preformed wells in the E. faecalis-covered MH agar plates that were then incubated overnight at 35°C. Finally, inhibition zones (in millimeters) surrounding the vancomycin/Peptide-1-containing wells were measured, for determination of vancomycin deactivation by Peptide-1. Wells containing vancomycin alone and Peptide-1 alone served as positive and negative controls, respectively.
Antagonizing enhancement of vancomycin antibacterial effect by modified peptides
To improve the affinity of Peptide-1 to vancomycin, two different moieties were attached to the Lys residue, replacing the acetyl groups. The insertion of each moiety yielded a set of three analogs replacing either acetyl or both, as shown in Table 1 and Figure 1.
The covalent attachment of the modified analogs to vancomycin was anticipated to be formed by replacement of the bromine or fluorine on the bromoacetyl or on the FSBA moieties of the modified tripeptides, respectively, with one of three hydroxyl groups on the tyrosine residues of vancomycin (in addition to the five hydrogen bonds formed between the a-a terminus and vancomycin). A set of experiments similar to the initial set described above was conducted using each of the six tripeptide analogs. The first modification involved replacing the acetyls with bromoacetyl (Petides 2–4) whereas the second modification involved the use of FSBA to replace the acetyls (Peptides 5–7).
Vancomycin:peptide-analog molar ratios tested were 2:1, 1:1, 1:5, and 1:30. To ascertain whether bromoacetic acid and FSBA alone antagonize vancomycin's antibacterial activity in absence of the tripeptide, two additional sets were prepared with vancomycin:bromoacetic acid/FSBA molar ratios of 2:1, 1:1, 1:5, and 1:30 (and appropriate controls), one incubated for 1 hour before being transferred to E. faecalis growing MH plates and the second incubated for 24 hours. Identical sets were prepared using E. faecium ATCC 35667 as the tested bacteria.
Prevention of in-vitro selection of resistant Enterococci from a mixed susceptible/resistant population following exposure to vancomycin
To determine whether antagonism of vancomycin activity by Peptides 5–7 can prevent selection of vancomycin-resistant bacteria, two Enterococci species were used: a vancomycin-susceptible, ampicillin-resistant E. faecium strain (a wild-type clinical strain obtained at the clinical microbiology laboratory, designated U-2121), and a vancomycin-resistant, ampicillin-susceptible E. faecalis strain (ATCC 51299). Susceptibilities and MIC values of the test strains, determined using E-test strips, are presented in Table 2.
MIC, minimal inibitory concentration.
The opposing susceptibility/resistance properties of these two strains in relation to vancomycin and ampicillin served as phenotypic markers. For each strain, a 6×108 CFU/ml suspension in sterile saline (equivalent to 2.0 McFarland; bacterial density determined using a McFarland densitometer) was prepared. Vancomycin solutions were 12 and 6 mg/L in sterile saline. A single FSBA tripeptide analog (Peptide-6) was used in this experiment. First, reaction solutions of 1:5 molar ratio of vancomycin:Peptide-6 were prepared for both vancomycin concentrations. Following a 1-hour incubation period at 35°C, bacterial suspension was added in a 1:1 volume ratio (thus decreasing both bacterial density and vancomycin concentration by one half, to approximately 1.0 McFarland and 6 and 3 mg/L, respectively). Bacterial suspensions were the ATCC-51299 strain, the U-2121 strain, or a 1:1 mixed population, prepared by mixing an equal volume of each strain. Following brief vortexing and spinning, reaction vessels were incubated for 18 hours at 35°C. Next, 10 mcl aliquots from each reaction vessel were evenly dispersed on a standard MH agar plate using a sterile swab. Two standard antibiotic susceptibility discs were placed on each plate: vancomycin 30 mcg and ampicillin 10 mcg (BBL Sensi-Disc; BD). Following incubation at 35°C for 24 hours, growth patterns upon each plate were observed and inhibition zones surrounding each disc were determined and recorded. An untreated sample of the mixed bacterial population (i.e., unexposed to either vancomycin or the FSBA-tripeptide analog) served as control.
Susceptibility/resistance indications derived from inhibition zone measurements encircling the ampicillin and vancomycin disks were based on Clinical and Laboratory Standards Institute
Results
Antagonism of vancomycin activity versus E. faecalis by Peptide-1
At Peptide-1:vancomycin molar ratios of 1:2, 1:1, and 5:1, vancomycin activity was not altered, resulting in inhibition zones of approximately 20–22 mm, similar to that of vancomycin alone. At a molar ratio of 10:1, a decreased inhibition zone of 17 mm was apparent, whereas at molar ratios of 28:1 and 56:1 no inhibition zone existed, similar to that of Peptide-1 alone. These results were obtained for both sets prepared, with and without the 1-hour preincubation period. Images of these results are shown in Figure 2.

Peptide-1 (Nα,Nε-diacetyl-tripeptide) versus vancomycin over a range of molar ratios, as determined by inhibition zones of ATCC 29212 Enterococcus faecalis growth on Mueller-Hinton agar plates.
Antagonism of vancomycin activity versus E. faecalis by bromoacetyl- and FSBA-tripetide analogs
Peptides 2–4 exhibited a weaker effect on vancomycin activity than the parent tripeptide (data not shown), suggesting that the insertion of the bromine interferes with the formation of hydrogen bonds essential for recognition and binding. Alternatively, the bromine moiety may cause steric hindrance effects that prevent the peptide from attaching to the binding site.
Antagonism of vancomycin activity by all three FSBA-tripeptide analogs (Peptides 5–7) was not evident at FSBA-tripeptide-analogs:vancomycin molar ratio of 0.5:1 that resulted in inhibition zones of approximately 19–20 mm, similar to that obtained with vancomycin alone. In a molar ratio of 1:1, a slight decrease in inhibition zone was evident (17–18 mm for Peptide-5 and Peptide-6, 13–14 mm for Peptide-7), suggesting partial antagonism of vancomycin activity. At molar ratios of 5:1 and above, all three analogs induced complete antagonism to vancomycin, as indicated by total absence of inhibition zone, similar to that seen with the vancomycin-free FSBA-tripeptide analogs alone. Images of the Peptide-5 set are presented in Figure 3. FSBA alone, not attached to the tripeptide, did not antagonize vancomycin activity following a 1-hour incubation, even at a molar excess of 30. Following a 24-hour incubation period, partial antagonism was noticed only at a molar excess of 30, as indicated by a decrease in inhibition zone from 20 mm to 14 mm (not shown).
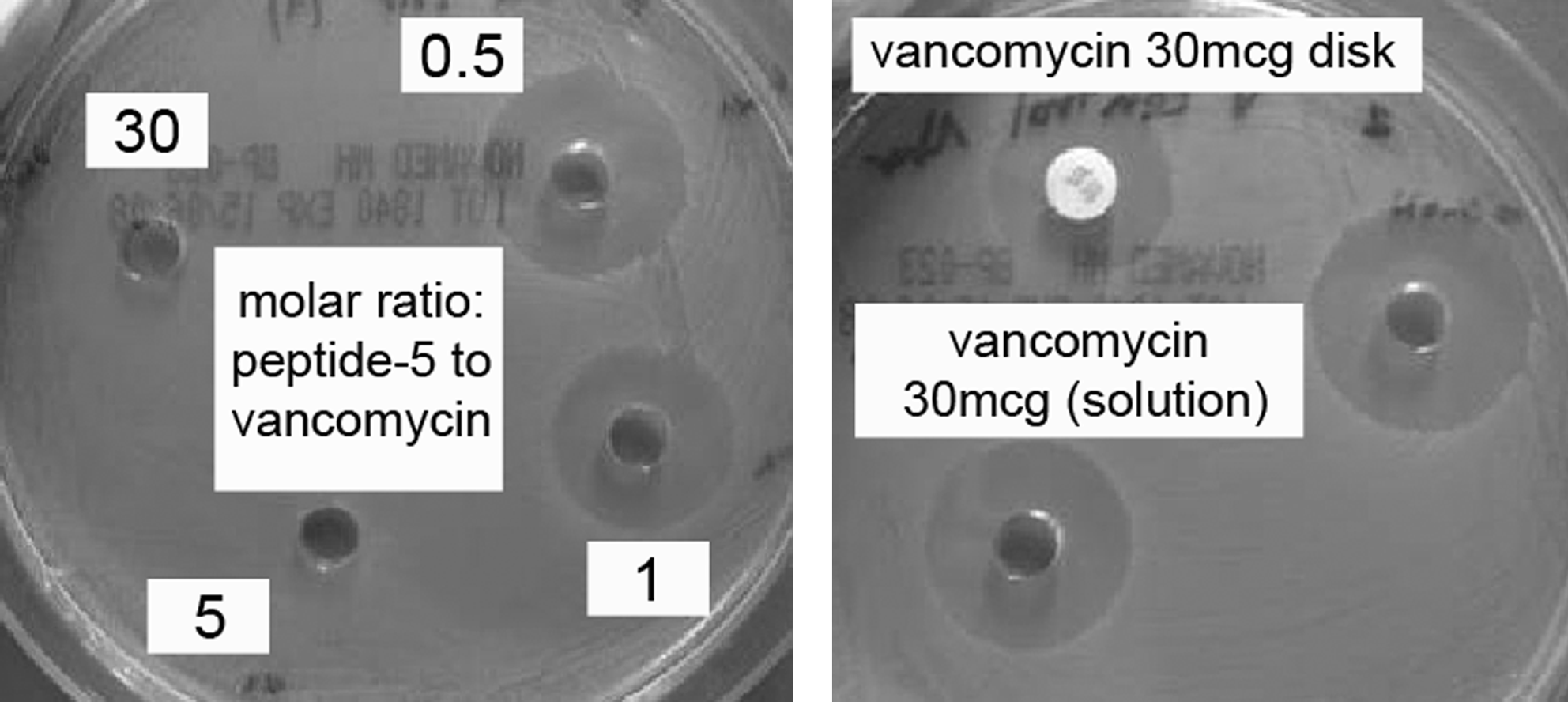
Peptide-5 (Nα-FSBA-Nε-acetyl-tripeptide) versus vancomycin over a range of molar ratios, as determined by inhibition zones of ATCC 29212 E. faecalis growth on Mueller-Hinton agar plates.
Prevention of selection of a VRE strain from a mixed population of susceptible/resistant strains following exposure to vancomycin by an FSBA-tripeptide analog
Vancomycin-susceptible, ampicillin-resistant U-2121 strain
Following exposure to vancomycin, at both concentrations tested, no bacterial growth was evident. Following exposure to the pretreated vancomycin-Peptide-6 solution, an inhibition zone of 25 mm (indicating susceptibility to vancomycin) was noted surrounding the vancomycin disk, and no inhibition zone was noticed surrounding the ampicillin disk. This was similar to the findings observed with the control sample, in line with full neutralization of vancomycin activity by Peptide-6.
Vancomycin-resistant, ampicillin susceptible ATCC-51299 strain
Following exposure to either vancomycin or vancomycin-Peptide-6 or no treatment (control), similar growth patterns were observed: a large inhibition zone (32 mm, indicating susceptibility) surrounding the ampicillin disk with two apparent inhibition zones (at 19 mm and at 10 mm) surrounding the vancomycin disk. This pattern is typical of VRE (the CLSI breakpoint for vancomycin resistance by Enterococci is 14 mm7). These results demonstrate the lack of effect of vancomycin activity (or lack of it due to neutralization by Peptide-6) on growth patterns of vancomycin-resistant strains.
Mixed 1:1 U-2121/ATCC-51299 population
The untreated control sample presented growth patterns consistent with the mixed properties. Surrounding the vancomycin disk, two inhibition zones were apparent: an 18 mm inhibition diameter, indicating presence of a vancomycin susceptible strain, and an inner inhibition diameter of 10 mm, indicative of copresence of a vancomycin-resistant population. Similarly, two inhibition zones were also noted surrounding the ampicillin disk; a 32 mm inhibition zone formed by an ampicillin-susceptible strain with a lower-density growth within this zone and reaching the disk, indicating copresence of an ampicillin-resistant strain. Following exposure to vancomycin, growth patterns of the mixed bacterial population resembled those of the ATCC-51299 strain alone (a 32 mm inhibition zone surrounding the ampicillin disk and an inner inhibition zone of 10 mm surrounding the vancomycin disk), a finding which is consistent with eradication of the vancomycin-susceptible strain and selection of the vancomycin-resistant strain from the initially- mixed population. On the other hand, pretreatment of the mixed population with vancomycin-Peptide-6 solution resulted in growth patterns similar to that of the control sample. This finding is consistent with neutralization of vancomycin activity and consequent preservation of the original bacterial population.
Inhibition zone diameters of all samples are summarized in Table 3.
Growth-inhibition zones on Mueller-hinton agar plates following exposure of 1.0 McFarland.
(1) vancomycin-susceptible, ampicillin-resistant Enterococcus faecium (VSE, U-2121); (2) vancomycin-resistant, ampicillin-susceptible Enterococcus faecalis (VRE, ATCC-51299), or (3) a mixed 1:1 population to (A) normal saline (control), (B) 3 mg/L vancomycin (vanc), or (C) 6 mg/L vancomycin following pretreatment with Peptide-6 at a molar ratio of 1:5 (vanc:Peptide-6).
Two inhibition zones are indicative of VRE growth.7
The 32 mm diameter inhibition refers to the VRE, being ampicillin-susceptible, whereas for VSE, being ampicillin-resistant, no inhibition was observed.
The 18 mm inhibition diameter refers to the VSE, whereas the 10 mm inhibition diameter refers to the VRE.
amp-R, ampicillin-resistant; amp-S, ampicillin-susceptible; Peptide-6, (Ac-Lys(FSBA)-a-a); Vanc, vancomycin.
Discussion
The expanding phenomenon of antimicrobial resistance is among the most troubling issues facing healthcare and public health worldwide in the 21st century. While the rate of development of new antimicrobials is consistently declining, emergence of resistant, multidrug resistant and pan-resistant strains of pathogenic bacteria is rapidly increasing. In this worrisome situation, effort should not be spared to preserve the clinical utility of existing antimicrobials. A strategy aimed at minimizing unnecessary exposure of bacteria to antibiotics may be productive in combating the emergence and spread of antimicrobial resistance.
In this paper we lay a foundation for one such strategy, devised to decrease one of the driving forces for selection of vancomycin resistance, a consequence of the unintentional and undesired encounter between the natural colonic microflora and vancomycin excreted via the biliary and fecal routes. Unrecognized until recently, the presence of significant amounts of parenterally-administered vancomycin in feces 9 may result in establishment and persistence of rectal colonization of vancomycin-resistant bacteria imposing an epidemiological threat to those in proximate contact with the carrier.
In the present study we demonstrated the loss of vancomycin antibacterial activity produced by trapping the antibiotic with a simple tripeptide derivative that mimics the natural bacterial binding site for this antibiotic within the bacterial cell wall. Further, we showed that enhancement of the basic tripeptide binding to vancomycin can be achieved with simple chemical modifications, resulting in deactivation of vancomycin at near-equimolar ratios. Attachment of a bromine group did not result in intensification of the tripeptide analog; however, linking an FSBA group decreased the tripeptide:vancomycin molar ratio required for neutralization of vancomycin activity from 28:1 with the parent tripeptide to less than 5:1. Stoichiometrically, covalent binding between vancomycin and the tripeptide is expected to result in a molar ratio of 1:1. The larger molar ratio required in our studies may suggest that the enhanced binding is caused by additional intermolecular interactions, such as aromatic-ring stacking. In any case, neither reaching a 1:1 molar ratio between vancomycin and the tripeptide nor covalently binding them is obligatory for neutralization of vancomycin activity. Rather, the ultimate goal is achieving selective, high-affinity nonreversible binding, as demonstrated in-vitro in the present study. The combined binding of vancomycin to both the FSBA moiety and to the tripeptide results in total annulment of vancomycin antibacterial activity against E. faecalis, a finding bearing significant potential benefit in both clinical and epidemiologic terms. The final study phase confirmed in-vitro that neutralization of vancomycin activity by an enhanced tripeptide analog can prevent selection of VRE from a mixed susceptible/resistant Enterococci population. Under selective pressure, in the form of exposure to vancomycin, the vancomycin-susceptible components of mixed susceptible/resistant bacterial populations are eradicated, thus selecting the resistant components. Pretreatment of vancomycin with the enhanced FSBA-tripeptide analog was proven to prevent this process, an attractive feature in combating vancomycin resistance.
The study presents several limitations. First, the study was conducted in-vitro. However, its findings merit further in-vivo exploration. Second, the chemical interactions between the enhanced tripeptide analog and vancomycin have not been characterized. Third, only two covalent labeling groups have been investigated, one of which (bromine) was found to be inactive. Additional chemical modifications and structure-activity relationship studies may yield analogs exhibiting improved binding properties. Finally, other aspects related to the enhanced tripeptide analog selection, such as selectivity and toxicity, were not studied. Transition to in-vivo studies will require careful consideration of complicating factors such as varying pH, nonspecific binding, intestinal peptide degradation, and facilitation of a timely encounter between the enhanced tripeptide analog and vancomycin. These factors will have to be taken into account in the design of the enhanced tripeptide analog and its delivery system.
In future studies complementing the present preliminary investigation, an enhanced tripeptide analog may be incorporated into an appropriately-designed pharmaceutical delivery system which, when administered orally according to a defined schedule relative to parenteral administration of vancomycin, will allow for trapping of the vancomycin portion excreted into the gastrointestinal tract via biliary excretion. As bile excretion in human occurs in response to presence of dietary fatty content in the stomach, 20 a pharmaceutical delivery system containing a fatty component may trigger such a response, thereby facilitating a synchronized encounter between biliary vancomycin and the enhanced tripeptide analog.
In conclusion, the novel strategy presented in this study offers a new prospect in the multipronged efforts for containing antimicrobial resistance. The present study should be viewed as a preliminary investigation, demonstrating the conceptual feasibility of the proposed approach. Importantly, the fundamental concept of neutralizing excess antimicrobials lacking therapeutic value while contributing to emergence and spread of antimicrobial resistance may be extended to other antimicrobial agents. Based on chemical, microbiological, pharmacological, and pharmacokinetic properties, suitable neutralization mechanisms may be identified and harnessed for provision of selective antimicrobial therapy and minimization of emergent antimicrobial resistance.
Footnotes
Disclosure Statement
No competing financial interests exist.