
Abstract
Traveler's diarrhea (TD) is an important public health concern that can result from a variety of intestinal pathogens, including bacteria, parasites, and virus. A number of antibiotics are being used to cure TD, but due to widespread use of these antibiotics, the pathogens are becoming resistant to them. In this work, we performed docking studies of DNA gyraseA (GyrA) and topoisomerase IV (ParC) of Shigella flexneri and their mutants with two different fluoroquinolones, ciprofloxacin and norfloxacin, to understand their resistance mechanism at the structural level. S. flexneri strains with mutations at serine 83 to leucine and aspartic acid 87 to glutamate or asparagine of GyrA and that of serine 80 to isoleucine in ParC have decreased susceptibility to fluoroquinolones. This analysis revealed that interaction of ciprofloxacin/norfloxacin with all the mutants was weaker than the interaction of ciprofloxacin/norfloxacin with the wild type. This study highlights the importance of aspartic acid and serine in GyrA and that of serine in ParC, forming bonds with ciprofloxacin/norfloxacin, which may play a crucial role in antibiotic resistance. This work corelates very well with the experimental outcomes and gives a good explanation for fluoroquinolone resistance in S. flexneri.
Introduction
D
A group of researchers working at the National Institute of Health Korea reported fluoroquinolone-resistant S. flexneri isolates from a stool sample of a patient who had traveled to India. 9 In that study, it was reported that in the susceptibility test of fluoroquinolone family antibiotics, S. flexneri (ATCC 29903) isolates with some mutations showed resistance to ciprofloxacin, norfloxacin, ofloxacin, and nalidixic acid whose minimal inhibitory concentrations are 8, 32, 8, and 256 μg/ml, 9 respectively. These mutations were Ser83→Leu and Asp87→Asn in GyrA and Ser80→Ile in ParC. A third mutation corresponding to Asp87→Gly in GyrA was also reported in another S. flexneri isolate from a Korean patient. 9 Similar mutations at the same positions were also studied in different Shigella strains and in serotypes such as S. flexneri 2a and 5a, Shigella sonnei, Shigella boydii, and Shigella dysenteriae.14,18 We studied the aforesaid mutations to investigate the resistance mechanism at the structural level.
In this work, we analyzed the molecular basis of the resistance mechanism by studying the interaction of two fluoroquinolones, ciprofloxacin and norfloxacin, with S. flexneri DNA gyrase A (GyrA) and topoisomerase IV (ParC) using molecular docking. Fluoroquinolones, one of the most effective second-line drugs, are antibacterial compounds used to treat various kinds of bacterial infections, including S. flexneri.16,23 These mainly target GyrA and ParC. 10 DNA gyrase plays an important role in the regulation of DNA topology, especially DNA super-coiling activity.16,20 DNA gyrase helps in the survival of bacteria inside the host cells. Topological stress that arises from the translocation of transcription and replication complexes along DNA is relieved by DNA gyrase, whereas topoisomerase IV being a decatenating enzyme resolves interlinked daughter chromosomes following DNA replication. 8 Emerging resistance to fluoroquinolones has been studied in several bacteria, such as in Staphylococcus aureus, Streptococcus pneumoniae, Pseudomonas aeruginosa, and Mycobacterium tuberculosis. 13 In the last few years, many studies related to the resistance mechanism have been reported, but structural level analysis revealing the mode of interaction of GyrA and ParC with fluoroquinolones needs to be explored. A recent study reported structural insights into the fluoroquinolone resistance mechanism of M. tuberculosis DNA gyrase A at the atomic level. 16 That study analyzed the functional, biophysical, and structural properties of the two individual domains constituting the catalytic DNA gyrase and thus identified original mechanistic properties of fluoroquinolone binding that represent relationships between amino acid mutations and resistance phenotype. 16 Likewise, we studied the resistance mechanism of S. flexneri GyrA and ParC by analyzing the structural alterations in the mutant protein relative to the wild-type protein using molecular docking studies. In some of the bacteria, such as S. flexneri and E. coli, GyrA and ParC act as the target for fluoroquinolones. 21 Therefore, these are suitable candidates for the study of the effect of mutations on fluoroquinolone resistance.
Materials and Methods
Here, the interaction study was carried out using LEADIT v2.1.6 package from BiosolveIT, 4 and Accelyrs Discovery Studio client 3.51 was used for molecule preparation.
Ligand preparation
The structures of the two ligand molecules ciprofloxacin and norfloxacin were taken from PubChem (https://pubchem.ncbi.nlm.nih.gov/), having identification numbers 2476 and 4359, respectively. The ligands were then prepared in Discovery Studio 1 and minimized by applying CHARMM force field and saved in MOL2 format for further use in docking studies.
Protein preparation
Homology modeling
The protein sequence of GyrA from the S. flexneri reference strains 2a (Accession number CEP59053.1) and 5a (Accession number EID62675.1), S. sonnei (Accession number CSP72916.1), S. boydii (Accession number WP_039060309.1), and S. dysenteriae (Accession number WP_001281279.1) showed 99% similarity to that from the S. flexneri (Accession number WP_001281258.1) isolate used in this study.
The X-ray crystal structure of the target proteins GyrA and ParC of S. flexneri was not available in the Protein Data Bank (PDB). The sequence of the proteins GyrA (Accession number WP_001281258.1) and ParC (Accession number KFZ98372.1) from S. flexneri was retrieved from NCBI protein database (www.ncbi.nlm.nih.gov). The sequence homology of S. flexneri DNA gyrase A and E. coli (strain K12) determined using NCBI BLAST 3 against the PDB database was about 98%, and that of S. flexneri ParC and E. coli was 99%, signifying that both the sequences are almost identical. E. coli K12 originated from a stool sample of a diarrhea patient had shown susceptibility to various drugs such as ampicillin, norfloxacin, ciprofloxacin, nalidixic acid, and erythromycin. 22 Also, similar mutations at the same positions in E. coli GyrA and ParC had shown resistance to two fluoroquinolones, ciprofloxacin and norfloxacin.11,17,23 Therefore, crystal structure of E. coli gyrase A (PDB_IDs: 1AB4) and E. coli ParC (PDB_ID: 1ZVU) was retrieved from PDB (www.rcsb.org) and was used as a template for homology modeling. The protein structure was then modeled using Accelrys Discovery Studio v3.5. The length of the protein sequence retrieved after modeling was 875 aa residues and 752 aa residues for GyrA and ParC, respectively. The best models chosen according to the lowest values of DOPE score were further evaluated using 3D verify (Accelrys Discovery Studio v3.5) 1 and ERRAT 5 program. This protein structure was referred in this study as wild type.
Mutated protein structure
Amino acid substitution corresponding to the mutation of Ser 83 to Leu and Asp 87 to Gly or Asn was introduced in the wild-type protein structure of GyrA using Accelrys Discovery Studio v3.5. 1 Since in our study we had taken only N terminal sequence of S. flexneri DNA gyrase A, the positions corresponding to mutations Ser 83 and Asp 87 in the modeled wild-type structure were residues 54 and 58, respectively. Hence, two mutated structures were generated: one structure having mutations at Ser 54 to Leu and Asp 58 to Asn was designated as mutant1, the other having mutations Ser 54 to Leu and Asp 58 to Gly was designated as mutant2.
Amino acid substitution corresponding to mutation Ser 80 to Ileu was introduced in the wild-type protein structure of ParC using Accelrys Discovery Studio v3.5. 1 This mutated structure was designated as ParC mutant.
Structure preparation and minimization
The wild and mutated protein structures were then prepared and energy minimized using Conjugate Gradient and CHARMM force field with a gradient of 0.1 in Accelrys Discovery Studio v3.5.
Molecular docking studies
After ensuring the correct conformations of protein and ligands, molecular docking of the ligands to the wild and mutated structures was performed using BioSolveIT (version 2.1.6) 4 FlexX algorithm. 12 The binding site consists of 40 amino acids starting from 31st residue to 70th residue for both wild-type and mutated molecules of S. flexneri gyrA, whereas in the case of ParC, the binding site consists of 30 amino acids starting from 61st residue to 90th residue for both wild and mutated molecules. The reason behind choosing this site is that it encompasses all the reported residues involved in the interaction of these antibiotics with GyrA and ParC of S. flexneri. A total of 100 structures with best poses based on score and hydrogen bonds were screened. A single best pose for each ligand was then chosen for further analysis.
Results
The results of ciprofloxacin and norfloxacin with respect to wild and mutated GyrA and ParC are discussed hereunder in terms of score and hydrogen bonds.
Ciprofloxacin binding with wild-type GyrA
The docking of ciprofloxacin with wild-type protein showed energy of “−21.8005 kcal/mol” (Table 1), involving five hydrogen bonds through residues Asp 58, Ser 68, Arg 62, and Gln 65 as shown in Figure 1. Also, the fluorine atom of ciprofloxacin made close contact with the residue Val 61 and also hydrophobically interacts with Asp 58. In general, a high docking score (more negative value) reflects a strong interaction between the ligand and protein molecule. The docking score here suggested a strong binding between target protein and ciprofloxacin.
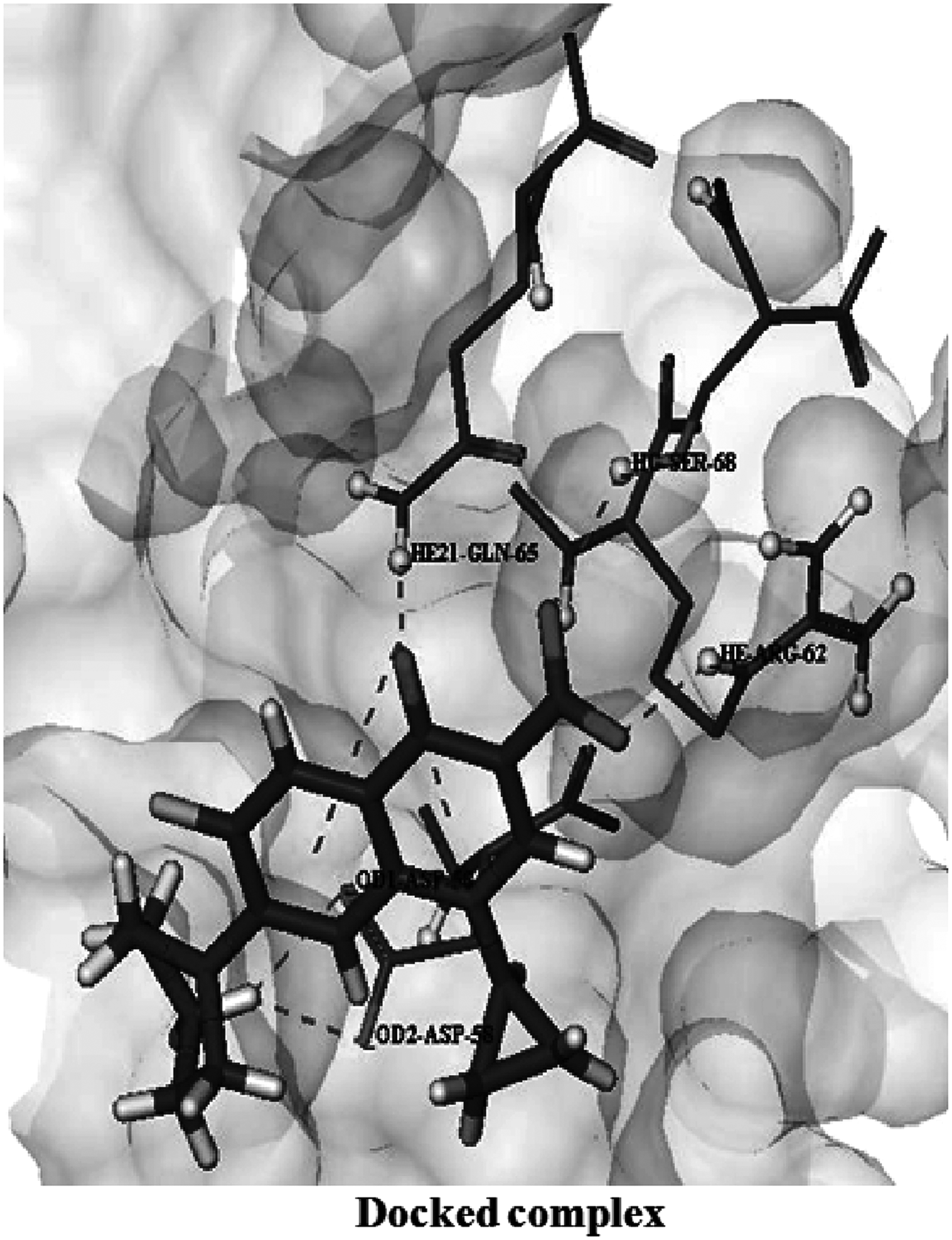
Interactions of ciprofloxacin with wild-type GyrA protein. Surface is presented in light gray. Ligand is shown in stick mode, GyrA interacting residues are shown in ball and stick model.
Ciprofloxacin binding with GyrA mutants
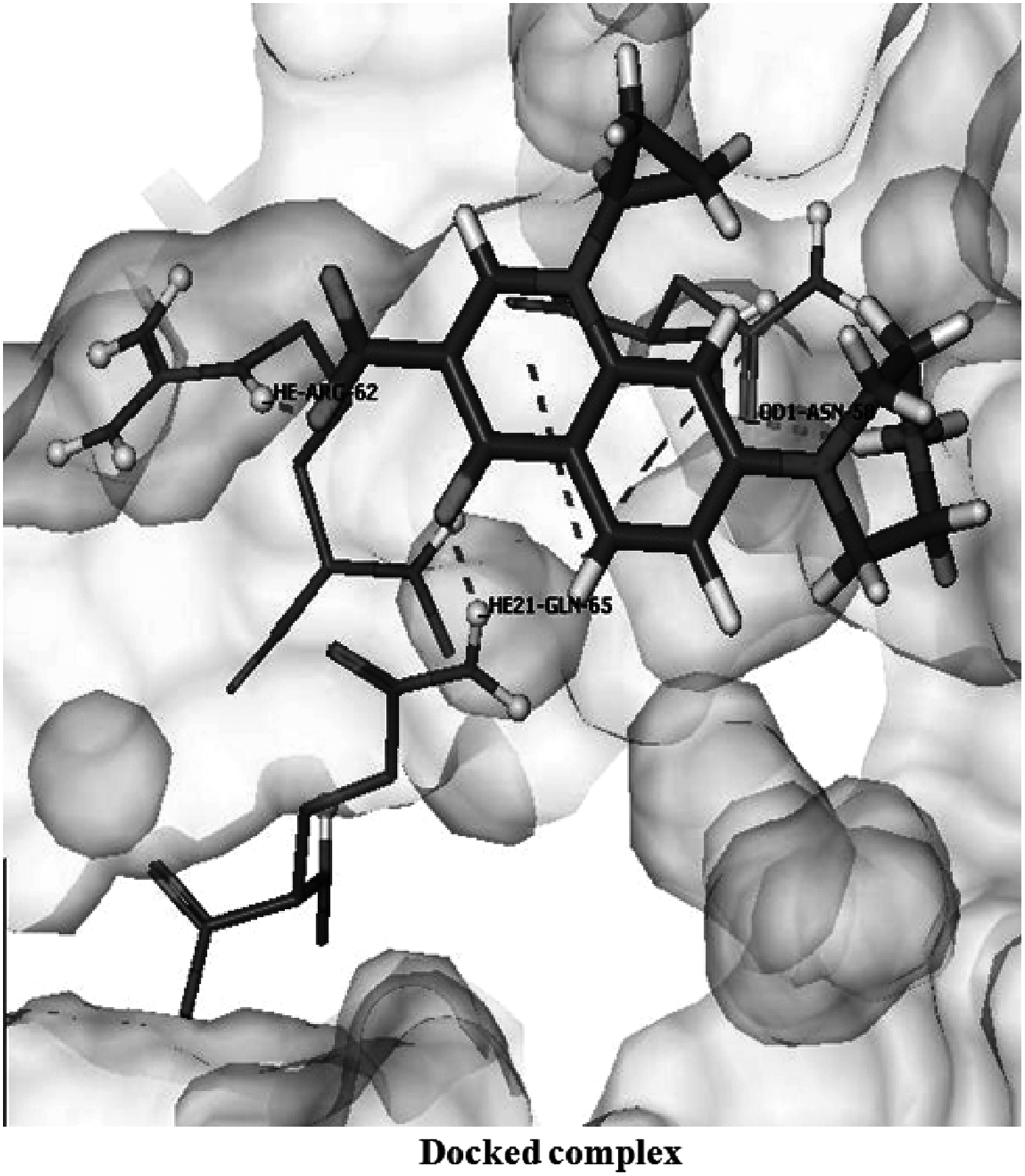
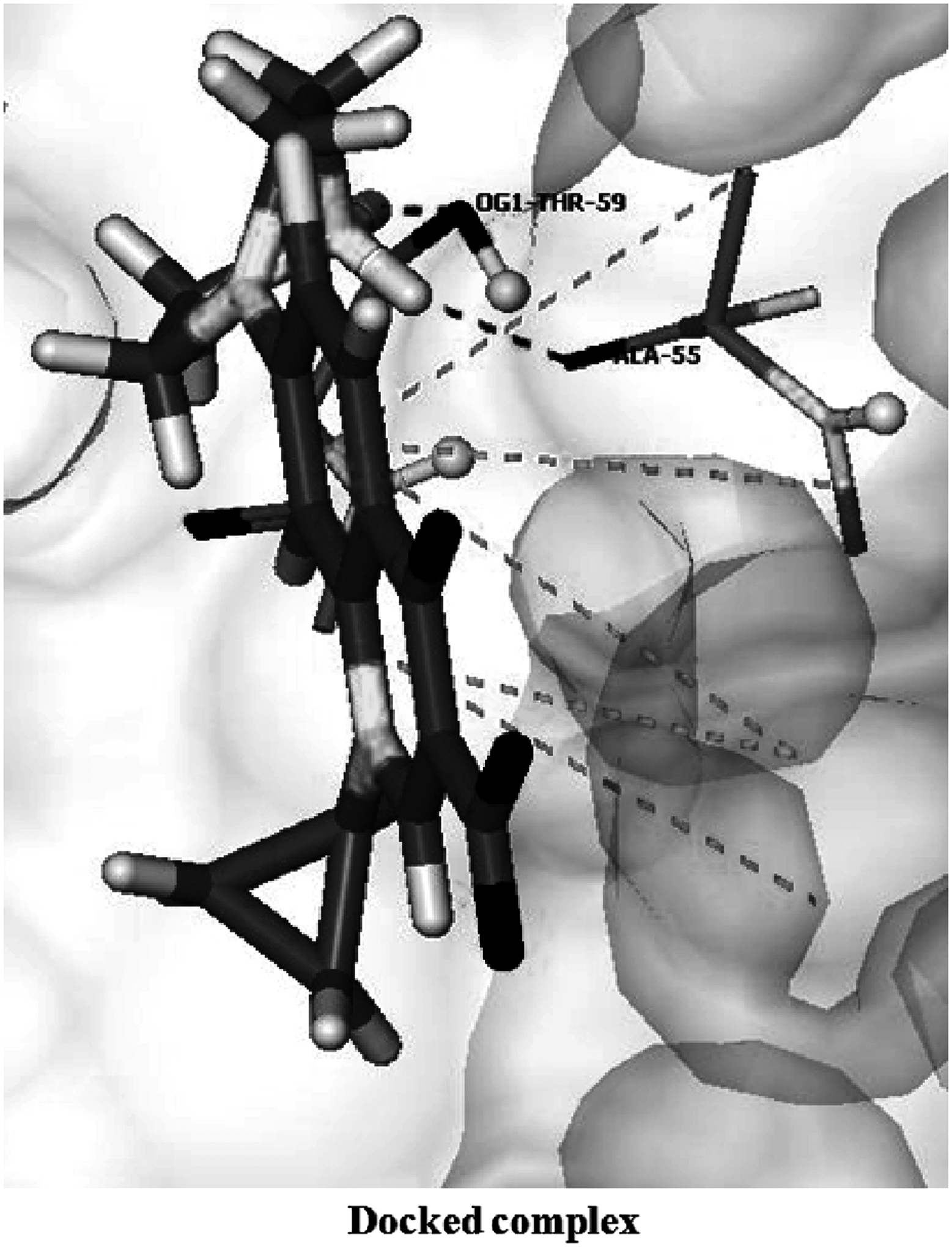
Decrease in the docking score was observed in both the structures, mutant1 (score = −18.3914 kcal/mol) and mutant2 (score = −17.2574 kcal/mol), involving only three bonds through residues Asp 58, Arg 62, and Gln 65 in case of mutant1, and two hydrogen bonds in the mutant2 structure, involving residues Ala 55 and Thr 59 (Figs. 2 and 3, respectively). Also, interacting amino acids in mutant2 were different from those in wild type. The major change noted here is the loss of two hydrogen bonds through residues Asp 58 and Ser 68, which showed the vital importance of these bonds in the binding of ciprofloxacin to GyrA of S. flexneri. Also, in this case, there is a considerable shortening of the hydrogen bond length (Table 1), which resulted in the distortion of some residues and their geometry.
Interactions of ciprofloxacin with mutant1 GyrA protein.
Interactions of ciprofloxacin with mutant2 GyrA protein.
Norfloxacin binding with wild-type GyrA
The docking of norfloxacin with wild-type protein showed a score of “−22.7593,” involving five hydrogen bonds through residues Asp 58, Ser 68, Arg 62, and Gln 65 (Fig. 4). This suggested a strong binding affinity between the target protein and the ligand molecule.
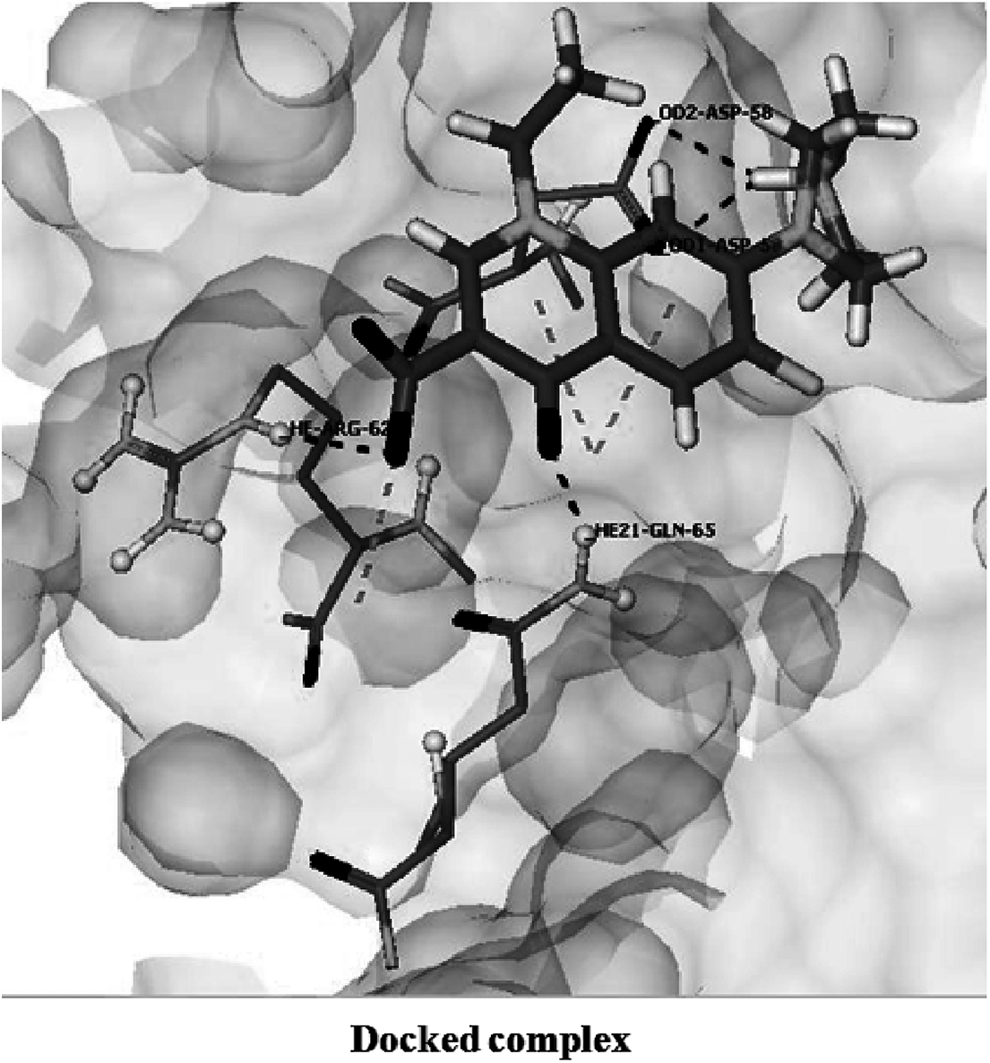
Interactions of norfloxacin with wild-type GyrA protein. Surface is presented in light gray. Ligand is shown in stick mode, GyrA interacting residues are shown in ball and stick model.
Norfloxacin binding with GyrA mutants
In this case also, it was observed that there is a considerable decrease in the docking score in both the mutated structures, mutant1 (score = −18.1598 kcal/mol) and mutant2 (score = −18.3914 kcal/mol) (Table 1), involving only three bonds through residues Asp 58, Arg 62, and Gln 65 in mutant1, and four hydrogen bonds in the mutant2 structure, involving residues leu 54, Arg 62, and Gln 65 (Figs. 5 and 6, respectively). Here also, in mutant1, hydrogen bond loss was observed, involving two residues Asp 58 and Ser 68. In case of mutant2, amino acids involved in interaction were different from those in wild type, and there are only four hydrogen bonds participating in this particular interaction. These lost hydrogen bonds might be of great importance and play a significant role in binding of these residues with norfloxacin. Here also, in mutant type, significant bond displacement was observed in some of the residues such as Asp 58 OD1 (where OD1 is the inner oxygen of the residue Asp forming hydrogen bond with the corresponding ligand molecule), Gln 65, and Arg 62 (Table 1).
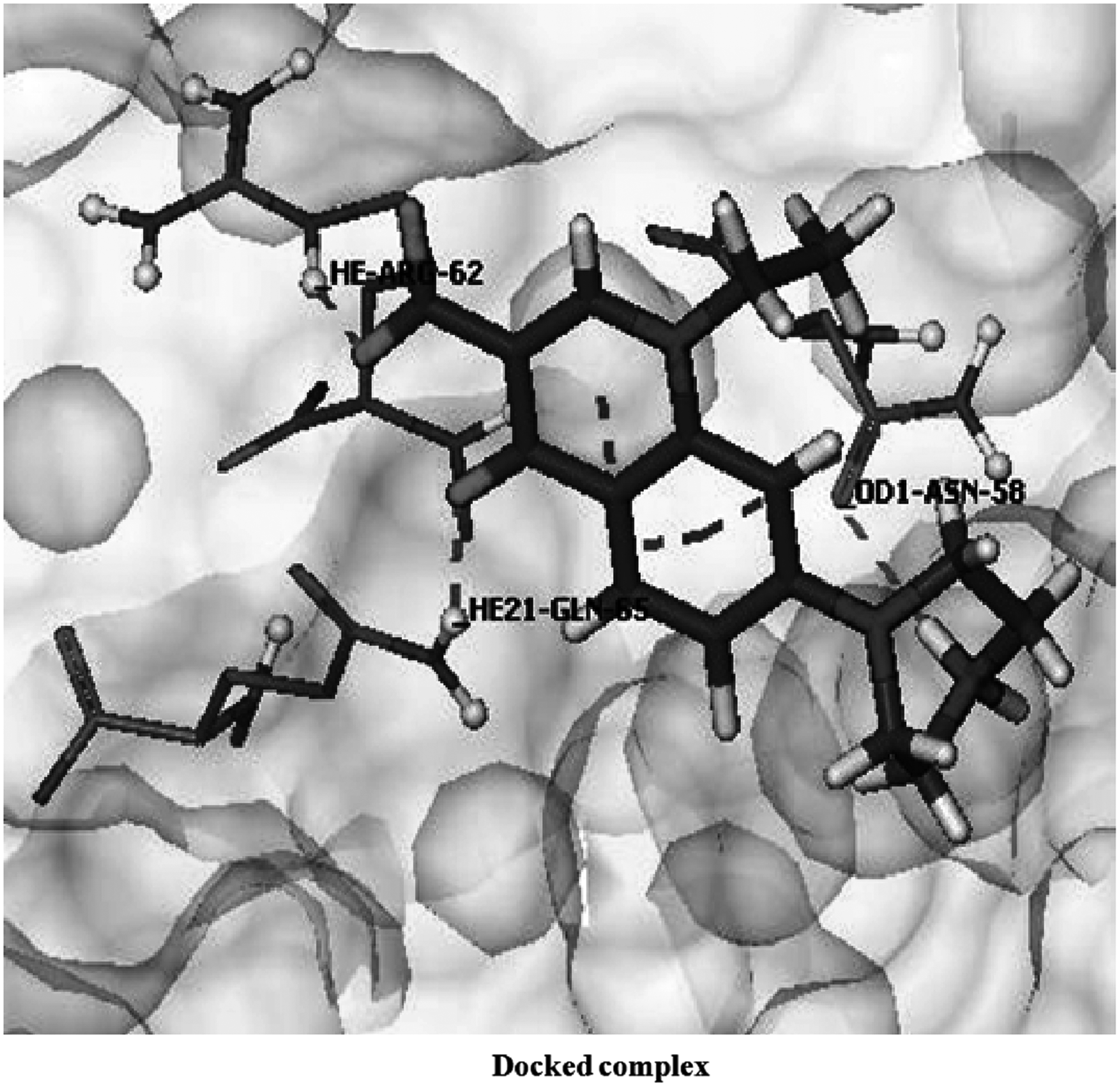
Interactions of norfloxacin with mutant1 GyrA protein.

Interactions of norfloxacin with mutant2 GyrA protein.
Ciprofloxacin binding with ParC
The docking of ciprofloxacin with wild-type protein showed energy of “−13.8863 kcal/mol” (Table 2), involving two hydrogen bonds through residues Ser 80 and Glu 84 as shown in Figure 7. The docking score suggested a strong binding between target protein and ciprofloxacin.
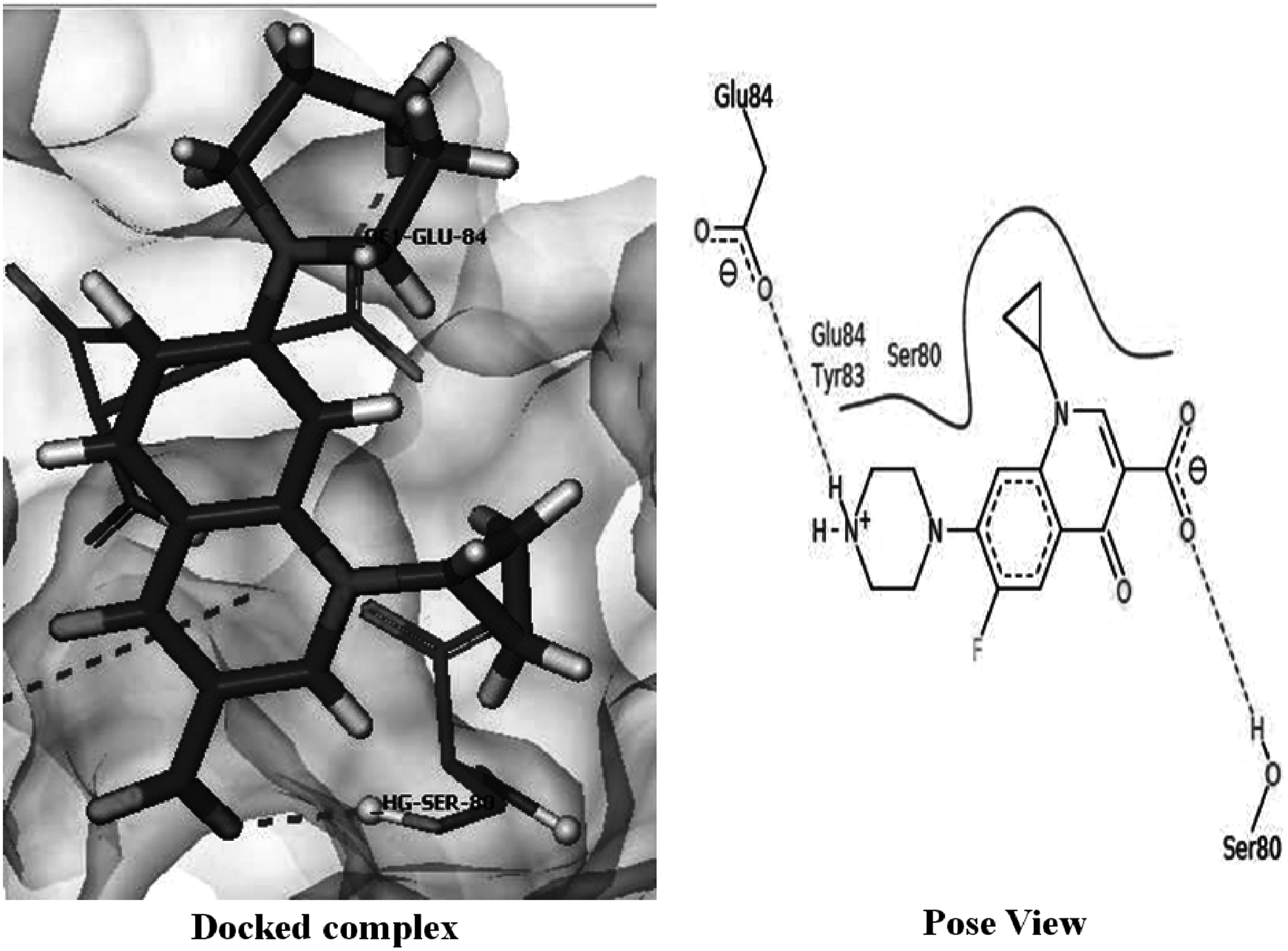
Interactions of ciprofloxacin with wild-type ParC protein.
After introducing mutations in the wild-type structure of ParC, remarkable decrease in the docking score was observed (score = −2.6835 kcal/mol), involving a single bond through residue Gly 78 (Fig. 8). It was observed that unlike Ser 80 residue, substituted Ile residues do not make direct hydrogen bond with ciprofloxacin and also there is increase in the hydrogen bond length in ParC mutant.
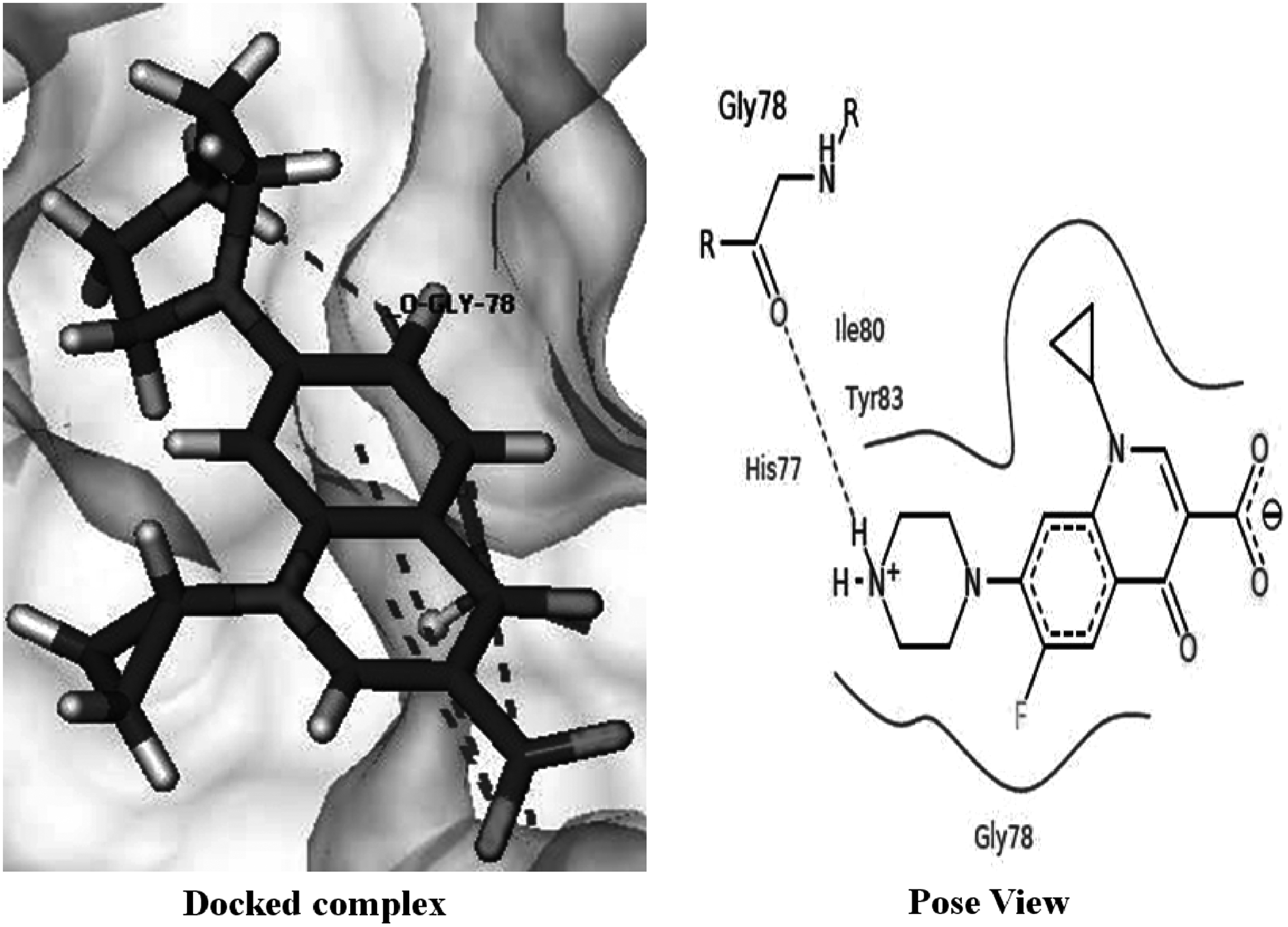
Interactions of ciprofloxacin with mutant ParC protein.
From these findings, it is clear that Ser 80 being directly hydrogen bonded to the ciprofloxacin in the wild-type protein plays a crucial role in the fluoroquinolone binding with ParC. This decreased docking score in mutant type accounts for increased resistance against fluoroquinolones.
Norfloxacin binding with ParC
The docking of norfloxacin with wild-type protein showed a score of “−11.4378 kcal/mol,” involving two hydrogen bonds through residues Ser 80 and Glu 84 (Fig. 9). This suggested a strong binding affinity between the target protein and the ligand molecule.

Interactions of norfloxacin with wild-type ParC protein.
Mutation of Ser 80 with Ile resulted in the considerable decreased docking score of −7.6866 kcal/mol, involving only one hydrogen bond through residue Ala 81 as shown in Figure 10. Loss of Ser 80 in mutant type suggests a vital importance of this residue in norfloxacin binding with ParC.

Interactions of norfloxacin with mutant ParC protein.
Discussion
The current computational studies provide insight into the interactions between target ligands (ciprofloxacin and norfloxacin) with S. flexneri GyrA and ParC and its mutants using molecular docking to calculate binding energies and identifying key residues participating in interactions. From the results obtained, it was evident that the decrease in docking score is more considerable in ParC than in GyrA. This decreasing order of binding suggested that mutations in ParC are more remarkable than mutations in GyrA, leading to induced resistance against the fluoroquinolones, ciprofloxacin and norfloxacin. This study reveals the relationship between the amino acid residues of the S. flexneri GyrA and ParC and the resistance mechanism to fluoroquinolones. Fluoroquinolone resistance takes place due to different mechanisms such as target site modification, by expulsion of the antimicrobial agents from the cell through general or specific efflux pumps or by plasmid-mediated fluoroquinolone resistance. Here, we have studied the resistance mechanism due to modification of the target binding site in bacteria. It was observed that both the mutations Ser 54 and Asp 58 in GyrA and Ser 80 in ParC are responsible for decreased interactions between fluoroquinolones, ciprofloxacin/norfloxacin, and of S. flexneri DNA gyrase A. The amino acid residues Asp 58 in GyrA and Ser 80 in ParC make direct hydrogen bonds with both ciprofloxacin and norfloxacin (wild type), so the mutations at this point lead to drastic changes in molecular interactions. The mutants have lower docking scores relative to the wild-type proteins. These are not only due to hydrogen bond but also due to hydrophobic interactions that take place between these fluoroquinolones and active site residues of S. flexneri. In case of mutation Ser 54 to Leu in GyrA, leucine being a bulkier molecule poses greater steric hindrance due to its side chain. From these findings, it is apparent that all the substitutions account for a decrease in the docking score and less efficient binding, ultimately leading to fluoroquinolone resistance in S. flexneri strains that were earlier sensitive to drugs.
Limitations of the study
The limitations to this study are the inherent limitations of the docking algorithm. Some of these are as follows: receptor flexibility, modeling cofactors, effectors, and solvation effects. 15 Hence, the docking algorithms need to be further improved in these directions for an increased reliability of the results.
Conclusion
The study presented here reveals the relationship between the amino acid residues of S. flexneri GyrA and ParC and their resistance mechanism to fluoroquinolones. All the substitutions are responsible for reduced affinity of these antibiotics against GyrA and ParC, which ultimately leads to resistance. The molecular docking studies showed good correlation with experimental studies, hence provide a possible explanation for antibiotic resistance in S. flexneri. Furthermore, these observations can be exploited to develop new drugs against the resistant strains of this pathogen.
Footnotes
Acknowledgment
We gratefully thank the Department of Science and Technology (DST), New Delhi, India, for awarding FAST TRACK Young Scientist Fellowship to J.R.
Disclosure Statement
This work was funded by the Department of Science and Technology (DST), New Delhi, India, and all authors report no other conflicts.