
Abstract
The aim of this study is to investigate the mutation pattern of the folC gene in drug-resistant Mycobacterium tuberculosis (MTB) clinical isolates of global and Hong Kong cohorts. The public sequence read archives of 1,124 MTB genomes from three independent studies were retrieved and folC mutations existing solely in drug-resistant MTB strains were identified. A phylogenetic tree was constructed to analyze the segregation of mutation-related amino acid residues in the FolC structure. These mutation sites were further supported by direct Sanger sequencing of the folC gene among 254 clinical MTB isolates in a Hong Kong cohort. Homology modeling of wild-type and mutated FolC was performed, and the predicted structures were docked with hydroxydihydropteroate, the metabolic derivative of para-aminosalicylic acid (PAS), to evaluate the resultant binding affinity changes. Combining the results of three previous cohorts and our cohort, E40, I43, S150, and E153 are the most frequently affected amino acid residues in resistant isolates. Based on the distribution of mutations in the genome-based phylogenetic tree, lineage-specific mutation patterns were observed. Regarding the segregation of affected amino acid residues, the four most frequently affected residues are all in close proximity of the binding pocket for the PAS derivative. Molecular modeling results showed that mutations at E40, I43, and S150 can alter the structure of FolC putative binding pocket, causing the PAS derivative to bind outside of the now deformed pocket. This might ablate the interaction between the protein and the PAS derivative. To conclude, this study is the first comprehensive mutation pattern and bioinformatics analysis of the folC gene in MTB drug-resistant isolates. The distribution of mutations in phylogenetic lineages and protein structure is reported, analyzed, and discussed.
Introduction
M
Rv2447c (folC) encodes for dihydrofolate synthase (FolC), a protein which is involved in the folate pathway of MTB. The synthetic pathway starts with the condensation between para-aminobenzoic acid and 6-hydroxymethyl-7,8-dihydropterin pyrophosphate to form 7,8-dihydropteroate using dihydropteroate synthase (FolP). FolC then catalyzes the glutamination of 7,8-dihydropteroate into dihydrofolate, which is further processed by dihydrofolate reductase to form tetrahydrofolate (THF). 2 THF serves as a ubiquitous one-carbon carrier for the metabolism of amino acids and nucleic acids. THF is also required for the biosynthesis of formyl-methionyl-tRNA, a key component for the initiation of protein synthesis in prokaryotes. Since the folate synthesis pathway is absent in human and in many mammals, enzymes involving in the folate pathway have been a frequent target for the development of antimicrobial agents.
Para-aminosalicylic acid (PAS) is an example of anti-TB agent that blocks the folate pathway in MTB. Due to the emergence of drug-resistant strains, PAS is currently being administered as part of the treatment regimen for some XDR-TB patients. Recent studies have delineated the detailed involvement of FolP in activating the drug through incorporating PAS with 6-hydroxymethyl-7,8-dihydropterin pyrophosphate to form hydroxydihydropteroate, a cytotoxic metabolite to MTB that inhibits the activity of downstream folate pathway and in turn arrests MTB growth. 2 Missense mutations at folC codons E40 and I43 have also been reported to confer high level of PAS resistance in spontaneous mutants. 3 However, the contribution of folC mutations to other antibiotic resistance mechanisms is still largely unknown. Further investigation is also needed to determine the PAS resistance-determining region in the FolC protein.
In this study, we focused on a collection of recent genome data from the public sequence read archives and identified mutations in folC gene. Combining with the folC mutations identified in the Hong Kong MTB cohort, we performed epidemiological and structural analyses on the mutation patterns and the corresponding functional significance. We found that different lineages and geographical regions have quite distinct patterns of mutations that may be attributable to clonal expansion. Moreover, the folC mutations exclusively identified in drug-resistant strains are clustered around the binding pocket of the FolC for hydroxydihydropteroate, and many of these mutations change the conformation of the binding pocket, leading to a significant reduction in putative hydrogen bonding between hydroxydihydropteroate and FolC.
Materials and Methods
Sequence read archives and retrieval of drug resistance profiles of three studies
A total of 1,124 MTB samples' raw genomic sequencing reads from three independent studies were downloaded from NCBI Sequence Read Archive (SRA) database and European Nucleotide Archive (ENA) database with the corresponding accession codes (SRA065095, SRA020129, SRA009637, SRA009341, SRA009458, and ERP000192).4–6 The drug resistance profiles of each clinical isolate were provided in the published articles. Samples from Farhat et al. were collected worldwide, whereas the samples from Casali et al. and Zhang et al. were collected from Russia and China, respectively. An MTB strain was classified as a drug-resistant strain if the sample is resistant to at least one of the four first-line antibiotics (RIF, INH, EMB, and STR) according to the drug profile provided.
Reference mapping of 1,124 genomes' raw sequencing reads and variant calling
All downloaded sequencing reads were mapped onto MTB H37Rv reference genome (Accession number: NC_000962.3) using Burrows-Wheeler Aligner (version 0.6.2). 7 All the variants from 1,124 genomes were called and concatenated into a single VCF file using SAMtools (version 0.1.19) 8 and GATK (version 3.0.0), 9 respectively. Clinical isolates that are resistant to either one of the four first-line antibiotics (isoniazid: INH; rifampicin: RIF; ethambutol: EMB; or streptomycin: STR) were further classified as strains carrying drug-resistant properties. SnpSift (version 3.4i) 10 was then applied to annotate genes and filter mutations that are present in drug-resistant strains. FolC mutations exclusively present in drug-resistant strains were selected, and resistant strains carrying these selected mutations were used for phylogenetic analysis and homology modeling.
Statistical correlation of folC mutation and the resistance profile to each antibiotic
All nonsynonymous mutations identified in folC were selected and passed to PROVEAN 11 to predict the functional deleteriousness caused by the missense mutation. Deleterious folC mutations were selected and chi-square test was performed to evaluate the statistical relationship between each mutation and the resistance profile to different antibiotics.
Phylogenetic analysis of clinical isolates
From the variant calling and filtering, we isolated 19 strains harboring folC nonsynonymous mutations, which are solely found in drug-resistant strains. The genomes of these selected strains, together with the reference genome H37Rv, were subjected to core-genome alignment and phylogenetic analysis using Parsnp (version 1.1) of the Harvest suite (version 1.0.1) using default parameters. 12 The phylogenetic tree was then passed to a Python script powered by ETE 13 to add drug profiles and mutation annotation.
Direct Sanger sequencing of the folC gene in an MTB cohort in Hong Kong
A total of 254 clinical MTB isolates (including 49 DR, 81 MDR, 9 XDR, and 115 drug-susceptible) were collected from the Tuberculosis Reference Laboratory of the Hong Kong Special Administrative Region. These isolates have well-characterized drug-resistant profiles as well as complete clinical records. All isolates were subjected to direct Sanger sequencing of the folC coding sequence with the four sequencing primers (folC_ORF_F1: 5′-AGGAAACCGAGCGCATCAC-3′, folC_ORF_R1: 5′-AGCAGCACCTCCATGACCTTC-3′, folC_ORF_F2: 5′-AGAAGGCGGGCATCATCAC-3′, folC_ORF_R2: 5′ AGTATCAACAGCACGGCCAGAC-3′).
Homology modeling and molecular docking
The wild-type amino acid sequence of the FolC from the wild-type MTB strain H37Rv (RCSB PDB ID: 2VOR) was chosen as template for homology modeling in SWISS-MODEL, 14 and each resistant strain's mutated FolC sequences were aligned to the template. The best model generated with the highest GMQE score (>0.95) was downloaded from the SWISS-MODEL server. The structure of hydroxydihydropteroate was constructed according to its structural analog 7,8-dihydropteroate (DHP) available in ZINC (ZINC30320891). 15 SwissDock was employed to first predict the approximate interacting sites between the wild-type FolC protein model and hydroxydihydropteroate. 16 Since SwissDock returns multiple putative binding regions for the ligand termed clusters, we, therefore, selected the cluster with the lowest estimated free energy ΔG. The estimated docking coordinates obtained from the wild-type FolC protein model by SwissDock were recorded. The coordinates were passed to iDock, 17 a molecular docking pipeline for assessing the binding affinity of ligands to target proteins. The iDock software implements the RF score, which takes into account free energy change upon binding and circumvents the need for modeling assumptions during scoring by using Random Forest, and dramatically improves accuracy with training set size, which is steadily increasing. 18 The docking results were visualized and rendered by the PyMOL Molecular Graphics System (version 1.7.1.7 Schrödinger). 19
Results
To analyze the mutation patterns in folC, we first collected the sequencing read archives of 1,124 MTB strains from three independent studies, together with the drug profile and the geographical origin for each strain. Of the 1,124 MTB strains collected in this study, 391 were classified as drug-sensitive strains, whereas the remaining 733 belong to drug-resistant strains. The sequencing reads for each isolate were mapped to the reference genome H37Rv (Accession No.: NC_000962.3), and all nonsynonymous mutations identified in folC were called. All mutations identified in folC are listed in Table 1. folC missense mutations were found to be a rare event in drug-resistant MTB strains. Missense folC mutations were observed in 89 out of 1,124 strains. Among the 89 strains with folC mutations, 63 strains (37 drug sensitive; 26 drug resistant) belonging to the Russian cohort had a common missense mutation A420V, suggesting that this mutation may only be a marker for a specific sublineage in the Russian cohort, but does not contribute to drug resistance. After excluding A420V and other mutations that were identified in drug-sensitive strains, 24 out of 773 drug-resistant strains were found to carry folC mutations.
(A) List of folC mutations solely found in drug-resistant clinical isolates. (B) List of folC mutations solely found in drug-susceptible clinical isolates. (C) folC mutations found in both drug-resistant and drug-susceptible clinical isolates.
Synonymous mutations (SYN) and Nonsynonymous mutations (NS).
We then used the drug profile and the resultant mutation list to determine the statistical correlation between folC nonsynonymous mutations and drug resistance properties. Thirteen folC mutations were selected, which were distributed among 24 MTB strains. Among all the drug profiles we obtained, resistance toward ethionamide (ETH) has demonstrated a statistically significant correlation with folC mutations (p < 0.05, OR = 11.36). Although a statistically significant correlation between folC mutations and ethambutol (EMB) resistance profile can also be established in this assay (p < 0.05), the odds ratio is 2.67, which is much smaller than the odds ratio from reference tests of confirmed EMB resistance-related genes and the corresponding drug profiles (Table 2).
From the public sequence archives, strains with nonsynonymous folC mutations, which were predicted to be deleterious, were tested against the resistance profiles of each antibiotic. Abbreviations of antibiotics are listed according to Figure 1.
Since PAS is seldom considered in common treatment regimens, PAS resistance was only tested in a limited number of strains. The insufficiency of PAS resistance profile has restrained the accurate measurement of correlation between PAS resistance and folC mutation by chi-square calculation. The resultant p-value and odds ratio, therefore, was not included in this test.
NA, data not available.
To further explore the relationship between lineages and folC mutations, we isolated 19 genomes with folC mutations solely present in drug-resistant strains. Together with the reference genome H37Rv, a total of 20 MTB genomes were subjected to core genome alignment, which was followed by phylogenetic tree construction using Parsnp. The phylogenetic tree, along with a mutation site annotation plot for each strain illustrating the mutations in FolC, is shown in Fig. 1. As shown in Fig. 1, mutations were mostly found in two regions along the protein sequence. The first region occurs from E40 to I43, while the other region occurs from D135 to E153. Variants harboring mutations at these two sites were dispersed in different lineages, indicating that these nonsynonymous mutations were not occurring as a result of lineage effect.

Phylogenetic analysis of the folC gene among three studies.
The sequence of folC gene from 254 MTB clinical isolates was analyzed using Sanger sequencing to consolidate our findings from the aforementioned 1,124 MTB genomes. From our clinical samples, a total of 22 folC mutations were detected exclusively in drug-resistant strains (Table 1). Among these 22 folC mutations, 21 of them were occurring at codons I43, S150, and E153. We then mapped all the affected amino acid residues to the three-dimensional structure of H37Rv-FolC (RCSB PDB ID: 2VOR) (Fig. 2). Interestingly, although E40, I43, D135, S150, and E153 are distant from each other in the primary sequences, these amino acid residues cluster in close proximity when the protein is folded in its tertiary structure.
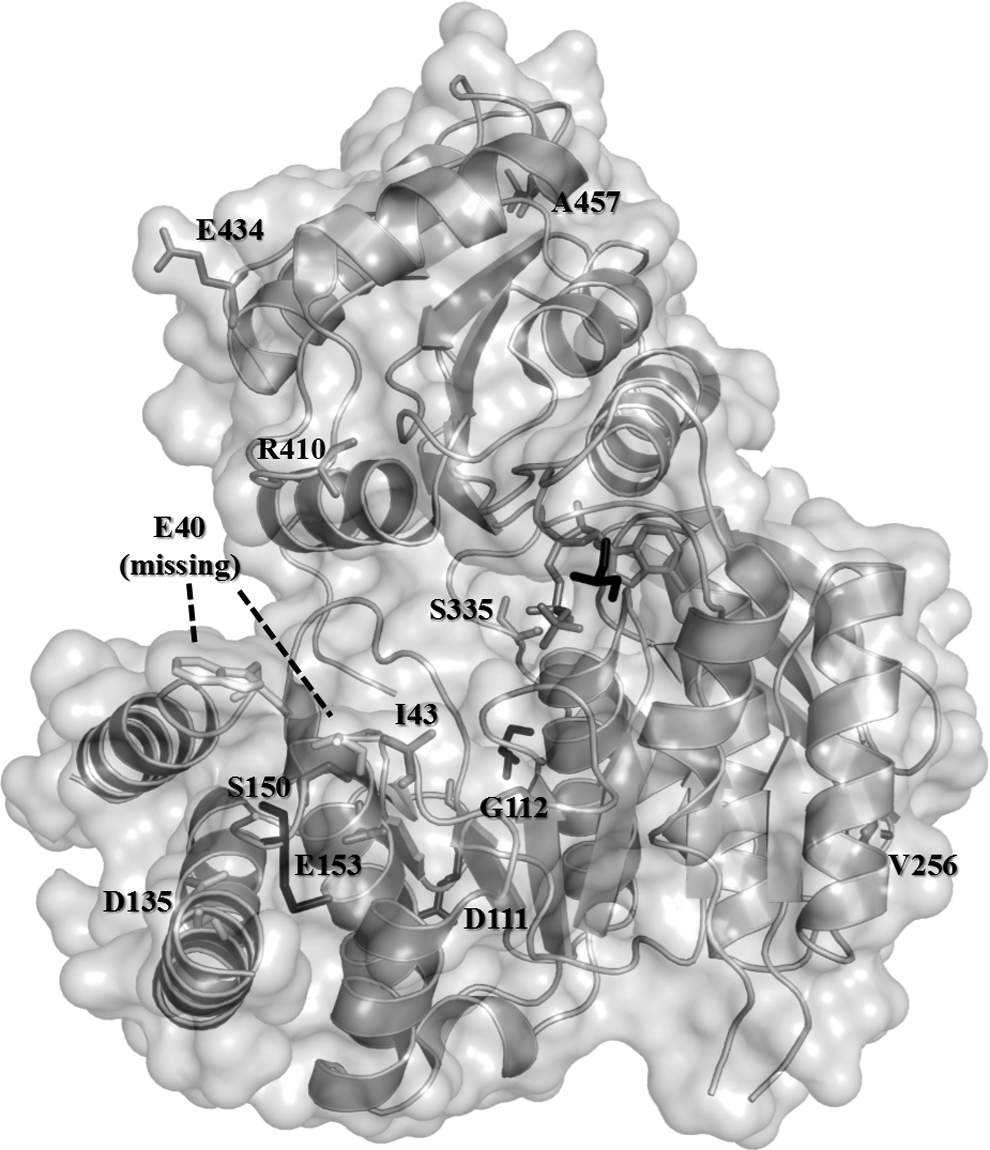
Mutation sites in three-dimensional FolC crystal structure. The MTB FolC structure was retrieved from the RCSB Protein Data Bank (PDB ID: 2VOR). Note that only nonsynonymous SNPs are presented in this figure using color coding. Black line molecule represents ACP, whereas iron gray molecule represents the PAS derivative. Since the location of E40 was absent from the X-ray crystal, its location and orientation with the lowest free energy (ΔG) was predicted by homology modeling.
To evaluate the structural consequences of the aforementioned mutations on the possible binding of hydroxydihydropteroate, we obtained all FolC mutant structures by homology modeling and then docked hydroxydihydropteroate into the FolC protein structures using iDock. The docking results of hydroxydihydropteroate with wild-type FolC and those binding pocket-related mutations (E40G, E40K, I43A, I43F, I43S, I43T, I43V, S150G, E153A, and E153G) are shown in Fig. 3. Since mutation A420V was identified in both drug-resistant and sensitive MTB strains, its docking result was regarded as a negative control.
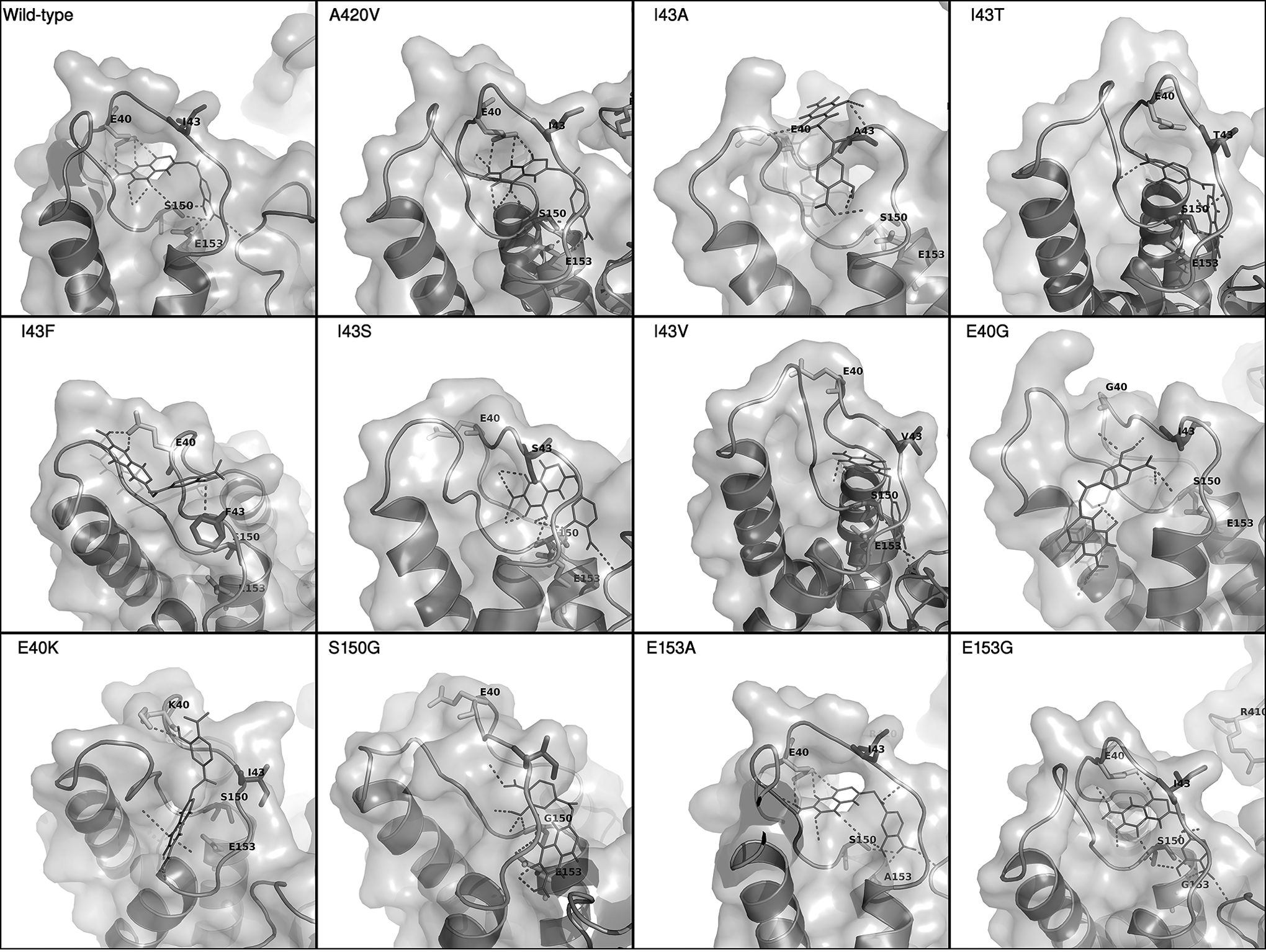
Molecular docking of the FolC mutants with the PAS derivative in the binding pocket. PAS derivative is indicated by the thin stick and the hydrogen bonds formed in the FolC binding pocket.
Discussion
In this study, we focused on the correlation between drug resistance properties and folC mutations. The thymidylate synthase (thyA) gene has been previously implicated in the PAS resistance. 20 Although we have shown that the positive predictive value of using thyA mutations for PAS resistance was 89.3%, the mutation frequency of this gene in PAS-resistant isolates is only approximately 36%. 21 Therefore, there is an urgent need to identify genes that can be used as alternative markers for PAS resistance. Recently folC mutations were also proved to be able to elevate the PAS resistance level in MTB. Drug susceptibility test showed that folC mutations at codon I43 were able to increase PAS resistance from 0.125 to 8 μg/ml. 2 Following this line of research, we performed a comprehensive study of folC mutations in drug-resistant MTB isolates. The information in this report will lay a solid foundation for subsequent research related to folC and PAS resistance.
It is notable that folC mutations were mostly clustered at 2 regions. The first cluster centers on codons E40 to I43 in the partially disordered DHP binding loop over the pteridine ring. 22 Whereas for the second cluster, it is located at codons D135 to E153 within the middle domain of the UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase protein family. 23 Interestingly, within these two clusters there are three residues that could be changed into various amino acids in drug-resistant strains, including E40, I43, and E153. This phenomenon could also be observed in other drug-resistant genes, including rpoB and gyrA, especially at residues that are essential for conferring antibiotic resistance. Among these three amino acid residues, I43 is the residue most likely to mutate in nearly all cohorts and could be changed into five different residues, with I43T being the most common form (9 out of 13).
When we investigated the affected amino acid residues in the FolC structure, most of them are located at the coil regions of the protein (9 out of 12 drug resistance-related amino acid residues in the coil regions compared with only 39% of the coil regions in the folC protein). It is possible that changes in the coil regions will have limited effect on the overall conformation of the protein, but the resultant structural alterations could still possess the ability to confer resistance to some extent. We then attempted to determine the docking region of hydroxydihydropteroate on wild-type FolC structure. The result showed that hydroxydihydropteroate can interact with the binding pocket formed by α1-α2 loop and α4-α5 loop, which is consistent with previous studies on the interacting site between hydroxydihydropteroate and FolC. Interestingly, the two aforementioned mutation clusters are also residing within these two binding loops, and the clusters are folded into close proximity despite being distant from each other in the primary sequence. This further indicates the importance of our selected mutations on PAS resistance properties.
In silico mutagenesis followed by docking illustrated that alterations at E40 and S150 abolish the interaction between hydroxydihydropteroate and FolC protein at the putative binding site, possibly due to the loss of hydrogen bond donor/receptor sites that are observed in wild type. Although no direct interaction was observed between the PAS derivative and residue I43, multiple mutations at this site might result in conformational changes that close the entrance to the binding pocket. This is consistent with the hypothesis that the drug-resistant mutations weaken the interaction between the PAS derivative and the FolC protein.
Missense mutations at I43A and I43T were reported to be positively correlated with PAS resistance in MTB clinical isolates. When comparing their docking results with the wild-type control, the mutants exhibited significant structural changes in the putative binding interface, leading to weakening of binding as determined in situ. Our docking results showed that the altered drug–protein interaction occurred outside the functional α1-α2 DHP binding loop, whereas the binding loop of the wild-type strain interacts more intimately with hydroxydihydropteroate. The shift of the binding interface from the pocket toward the protein surface leads to a significant reduction in putative hydrogen bonding between hydroxydihydropteroate and FolC. Similar results were identified in mutants E40G, E40K, I43F, I43S, I43V, and S150G. E153A is a common mutation observed in the Hong Kong drug-resistant cohort, but only minor differences in the number of putative hydrogen bonds were identified between the docking result of E153A mutant and that of wild type. The former was predicted to form hydrogen bond with the PAS derivative by the amino group at the backbone of the new alanine residue in lieu of the carboxyl group of the original glutamate side chain. Further investigation is needed to elucidate the effect of this minor shift in a single hydrogen bond.
Additional chi-square test has demonstrated an interesting statistical association between folC mutations and resistance toward ETH. So far, there is no experimental evidence to support the positive correlation of ETH resistance conferred by folC mutations, but previous studies have shown that a synthetic INH-NADP adduct has the ability to inhibit the activity of dihydrofolate reductase, 24 an enzyme which is also involved in folate synthesis. Being a drug that shares a number of common inhibitory mechanisms with INH, whether mutations in folC can confer a certain level of ETH resistance in MTB is also worth investigating.
Footnotes
Disclosure Statement
No competing financial interests exist.