
Abstract
Salmonella Enteritidis has emerged as a global concern regarding quinolone resistance and invasive potential. Although quinolone-resistant S. Enteritidis has been observed with high frequency in Thailand, information on the mechanism of resistance acquisition is limited. To elucidate the mechanism, a total of 158 clinical isolates of nalidixic acid (NAL)-resistant S. Enteritidis were collected throughout Thailand, and the quinolone resistance determinants were investigated in the context of resistance levels to NAL, norfloxacin (NOR), and ciprofloxacin (CIP). The analysis of point mutations in type II topoisomerase genes and the detection of plasmid-mediated quinolone resistance genes showed that all but two harbored a gyrA mutation, the qnrS1 gene, or both. The most commonly affected codon in mutant gyrA was 87, followed by 83. Double codon mutation in gyrA was found in an isolate with high-level resistance to NAL, NOR, and CIP. A new mutation causing serine to isoleucine substitution at codon 83 was identified in eight isolates. In addition to eighteen qnrS1-carrying isolates showing nontypical quinolone resistance, one carrying both the qnrS1 gene and a gyrA mutation also showed a high level of resistance. Genotyping by multilocus variable number of tandem repeat analysis suggested a possible clonal expansion of NAL-resistant strains nationwide. Our data suggested that NAL-resistant isolates with single quinolone resistance determinant may potentially become fluoroquinolone resistant by acquiring secondary determinants. Restricted therapeutic and farming usage of quinolones is strongly recommended to prevent the emergence of fluoroquinolone-resistant isolates.
Introduction
N
In Salmonella, quinolone resistance is mainly mediated by point mutations of topoisomerase genes in the quinolone resistance-determining region (QRDR), usually in gyrA, occasionally in gyrB or parC, and rarely in parE. 7 Mutations causing amino acid substitutions in DNA gyrase subunit A (GyrA) of Salmonella have been commonly reported at codons 83 and 87 with different frequencies in serovars 8 and geographical areas. 9
Quinolone resistance in Salmonella can also be acquired by a number of plasmid-mediated quinolone resistance (PMQR) determinants, including the qnr genes, aac(6′)-lb-cr, qepA, and oqxAB.10,11 Of these, qnr genes encode a family of proteins with pentapeptide repeat that can bind to DNA gyrase and protect it by preventing an interaction with quinolones. 11 At present, qnr gene variants, qnrA, qnrB, qnrD, qnrS, and oqxAB, have been reported in nontyphoidal Salmonella worldwide.12–19
In Thailand, an increasing trend of quinolone resistance in S. Enteritidis was reported during the period 2001–2008.2,20 However, the QRDR mutational status and the prevalence of qnr genes in the serovars have not been elucidated. Therefore, this study aimed to investigate these quinolone resistance determinants in clinical NAL-resistant S. Enteritidis isolates collected nationwide in Thailand from 2004 to 2007. In addition, the relationship between these resistance determinants and multilocus variable number tandem repeat analysis (MLVA) typing results was analyzed to elucidate the dissemination of specific clones.
Materials and Methods
Salmonella strains
A total of 190 S. Enteritidis clinical isolates recovered from randomly selected blood (n = 99) and stool (n = 91) specimens were used in this study. The isolates were collected in regions throughout Thailand, namely Bangkok (n = 62), Northern (n = 32), Northeastern (n = 27), Eastern (n = 9), Central (n = 12), Western (n = 28), and Southern (n = 20) provinces by the WHO National Salmonella and Shigella Center, Ministry of Public Health, Thailand, during the period 2004–2007. Isolates were first categorized into susceptible, intermediately, or fully resistant to NAL, CIP, and NOR (Fig. 1) by the Clinical and Laboratory Standard Institute (CLSI) zone size criteria. 21
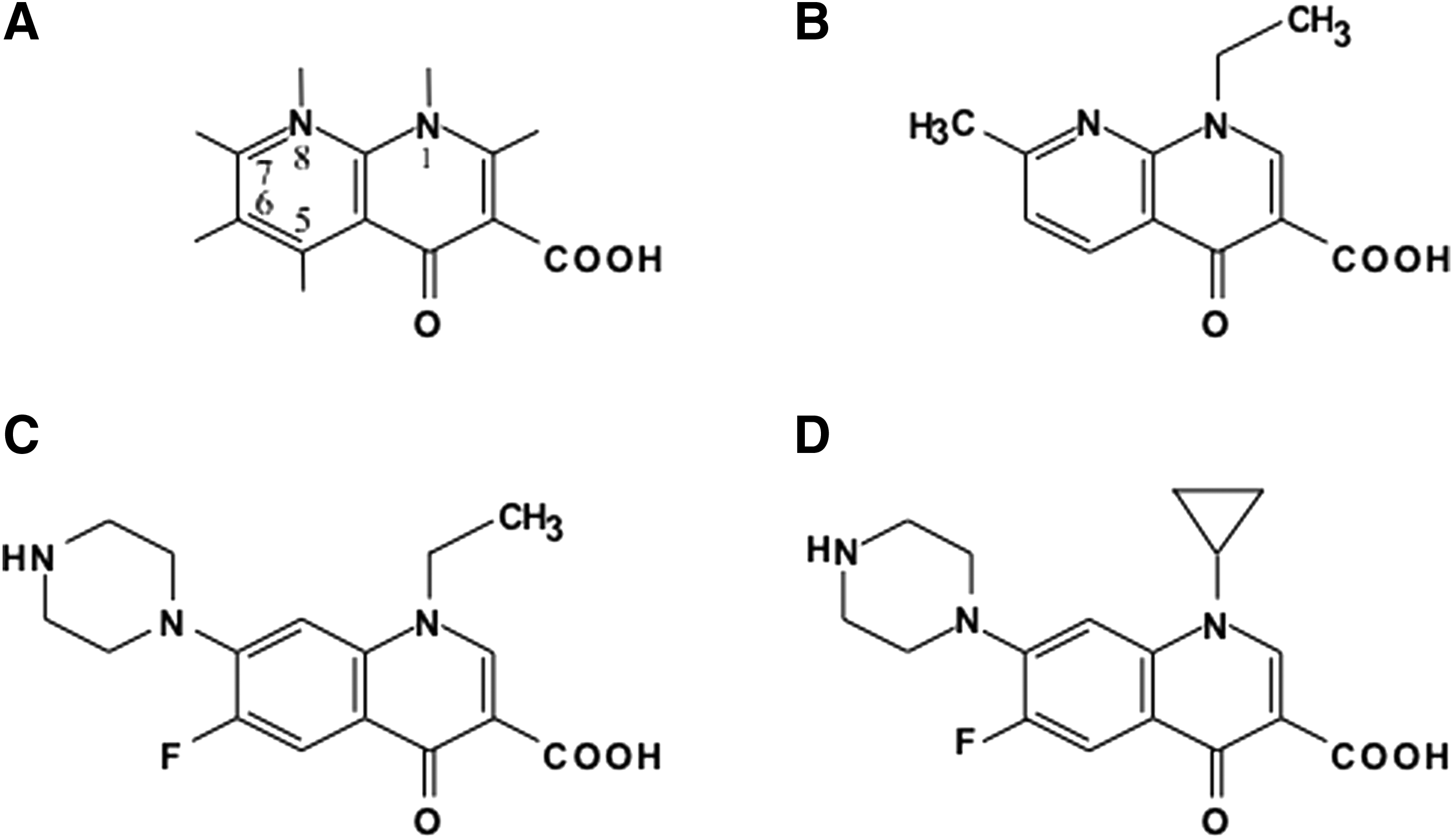
Structure of quinolones.
Determination of minimum inhibitory concentrations
The minimum inhibitory concentrations (MICs) of NAL, NOR, and CIP against intermediately or fully resistant strains by CLSI zone size criteria were determined by E-test (Biomerieux SA) according to the manufacturer's protocol. The clinical breakpoint used in this study was NAL, resistant (R) ≥32 mg/L, susceptible (S) ≤16 mg/L; NOR, R ≥ 6 mg/L, S ≤ 4 mg/L; and CIP, R ≥ 1 mg/L, S ≤ 0.06 mg/L, according to the CLSI guideline. 21 Escherichia coli ATCC 25922 was used as the quality control organism.
Detection of topoisomerase gene mutations
Genomic DNA was extracted using the DNeasy® Blood and Tissue Kit (Qiagen, Inc.) in accordance with the manufacturer's protocol. Mutations in the QRDR of topoisomerase genes gyrA, gyrB, and parC were analyzed by PCR and sequencing. We did not analyze parE as no previous report showed mutations in parE in quinolone-resistant Salmonella.
Three primer sets were designed to amplify these QRDRs (Table 1). The PCR mixture (20 μl) contained 1 μl of DNA template (1–10 ng), 0.25 mM dNTP (Biolabs, Inc.), 0.25 μM of each primer, and 1 U of GoTaq® DNA polymerase in 1 × Green GoTaq® buffer (Promega Madison). The amplification was conducted in an iCycler (BioRad) under the following conditions: predenaturation at 96°C for 1 min; 35 cycles of denaturation at 96°C for 10 sec; annealing at 52°C for 10 sec and extension at 72°C for 30 sec; and a final extension at 72°C for 5 min. DNA sequences of amplified fragments were analyzed using a BigDye® terminator, version 3.1, cycle sequence reaction kit and an ABI 3130 genetic analyzer (Applied Biosystems) in accordance with the manufacturer's instructions. Obtained sequences were compared with that of the reference strain P125109 with accession number NC_011294 in the GenBank.
QRDR, quinolone resistance-determining region.
PCR amplification and sequence confirmation of qnr genes
As qnrA, qnrB, and qnrS have been reported to be widely spreading, 11 the presence of these genes in isolates was examined by PCR using the primer sets designed by Robicsek et al. 22 under the amplification conditions used in their study. DNA sequences of amplicons were analyzed as above.
Characterization of plasmid carrying qnrS
Plasmids were purified from S. Enteritidis strain by NucleoSpin® plasmid kit (Macherey-Nagel) and E. coli Top10 (Thermo Fisher Scientific, Inc.) was transformed according to the manufacturer's manual. Colonies grown on the Luria-Bertani broth (Thermo Fisher Scientific, Inc.) containing 0.1 μg/ml CIP were selected for further analysis. Plasmids in transformants were identified using nuclease S1 digestion before PFGE, as previously described. 23
Briefly, DNA embedded in agarose was digested with 0.1 U/ml nuclease S1 (Promega) and separated by 1% agarose gels in a CHEF DR-III system. Electrophoresis was carried out at 6 V/cm at an angle of 120° with pulses ramping 30 sec for 13 hr in 0.5 × Tris-borate-EDTA buffer at 14°C. Southern blot hybridizations were performed by standard methods. 23 To determine the location of qnrS on plasmids, specific probes labeled with digoxigenin using the PCR DIG Labeling Mix (Roche Diagnostics GmbH) were used. Hybridization procedures and conditions were performed according to the manufacturer's manual.
Conjugation experiments were done among S. Enteritidis harboring qnrS-positive strains and E. coli Top10 or DH5α (Thermo Fisher Scientific Inc.) as recipients by mating on Luria-Bertani (LB) agar plate. Transconjugants were selected on LB agar containing streptomycin (100 μg/ml) + CIP (0.1 μg/ml) and confirmed by indole positivity as a marker for E. coli. Antimicrobial resistance profiles of S. Enteritidis isolates and transconjugants were determined by E-test or disc diffusion method in accordance with CLSI interpretation.
Multilocus variable number tandem repeat analysis
MLVA typing was conducted using previously designed primers for PCR amplification of five VNTR loci, SE1, SE2, SE5, SE7, and SE9,24,25 with some modifications (Table 2). The number of repeat units in locus SE7 was calculated from the amplicon size estimated by electrophoresis. Short repeat loci (SE1, SE2, SE5, SE9) were simultaneously amplified using four primer sets with fluorescence-labeled forward primers, and each product size was determined by capillary electrophoresis as previously described. The product size of each locus was measured and converted into the number of repeat units by Peak Scanner software (Applied Biosystems).
Sequence of reverse primers (SE1, SE2, SE5, SE9) were modified by adding nucleotides at the 5′ end for clear peaks by capillary electrophoresis.
VNTR, variable number tandem repeat; MLVA, multilocus VNTR analysis.
Antimicrobial resistance phenotypes
Antimicrobial resistance of 190 S. Enteritidis isolates to eight drugs, amoxicillin clavulanic acid (AMC), ampicillin (AMP), cefotaxime (CTX), chloramphenicol (CHL), gentamicin (GEN), streptomycin (STR), tetracycline (TET), and trimethoprim/sulfamethoxazole (SXT), was determined by the disc diffusion method according to the CLSI guideline. 21
Results
Quinolone resistance phenotypes
The disc diffusion test following the CLSI zone size criteria demonstrated that 158 isolates were intermediately or fully resistant and remaining 32 isolates were susceptible to all of NAL, CIP, and NOR. The distribution of MICs in 158 S. Enteritidis isolates with intermediate or full resistance to NAL is shown in Table 3. A high MIC of NAL (≥ 256 mg/L) was observed for 87.3% (138/158) of the isolates, while the remaining 20 isolates presented MICs ranging 16–64 mg/L. In addition, 143 isolates (90.5%) were NAL resistant (NALR). Except for one, all isolates were susceptible to NOR. As for CIP, 145 isolates (91.8%) had decreased susceptibility (CIPDS, 0.064 < MIC <1 mg/L), while only three isolates (1.9%) were resistant (CIPR). The most common phenotype, NALRCIPDS, was observed in 82.3% (130/158) of the isolates (Table 3).
MIC, minimum inhibitory concentration; CIP, ciprofloxacin; NAL, nalidixic acid; NOR, norfloxacin.
Topoisomerase gene mutations and presence of qnr genes
The sequencing results of gyrA, gyrB, and parC demonstrated that 138 of the studied isolates presented mutations in the QRDR of gyrA, while no isolates had mutations in either gyrB or parC. Mutations in gyrA were observed at codons 83 and 87, with three distinct amino acid substitutions in each codon (Table 3). Mutations appeared more frequently at codon 87 (87/138, 63.0%) than at codon 83 (50/138, 36.2%). A mutation causing an amino acid substitution of aspartic acid to tyrosine at position 87 (Asp87Tyr) was most frequently identified (83/138, 60.1%), followed by Ser83Tyr (40/138, 29.0%).
Eight isolates had mutations at codon 83, serine (T
Characterization of plasmids in qnrS1-positive S. Enteritidis isolates
A total of nineteen isolates were identified as positive for qnrS1 by PCR and sequencing (Table 4). Transformation experiments using extracted plasmids from S. Enteritidis strains positive for qnrS were performed using E. coli Top 10 as a recipient and all experiments gave CIP-resistant transformants.
AMC, amoxicillin + clavulanic acid; AMP, ampicillin; STR, streptomycin; TET, tetracycline; BK, Bangkok; C, Central provinces; E, Eastern provinces; N, Northern provinces; NE, Northeastern provinces; S, Southern provinces; W, Western provinces.
Then, S1 nuclease analyses using PFGE, followed by southern blotting, were done to show the size of plasmids to be 104.7 kbp and 52.7 kbp in strain ID134 and others, respectively (Fig. 2). Transconjugation experiments using the representative strains, SE-50 and −110, demonstrated the increase of MICs. The MICs of CIP and NOR against recipient E. coli Top 10 were <0.002 and <0.016 mg/L, respectively, whereas those against transconjugants with SE-50 were 0.125 and 0.5 mg/L, respectively. Similar increase of MICs of CIP and NOR was observed with transconjugant between E. coli DH5α and SE-110. Transconjugants also showed the reduction of the zone of inhibition by NAL and AMP (Table 5).
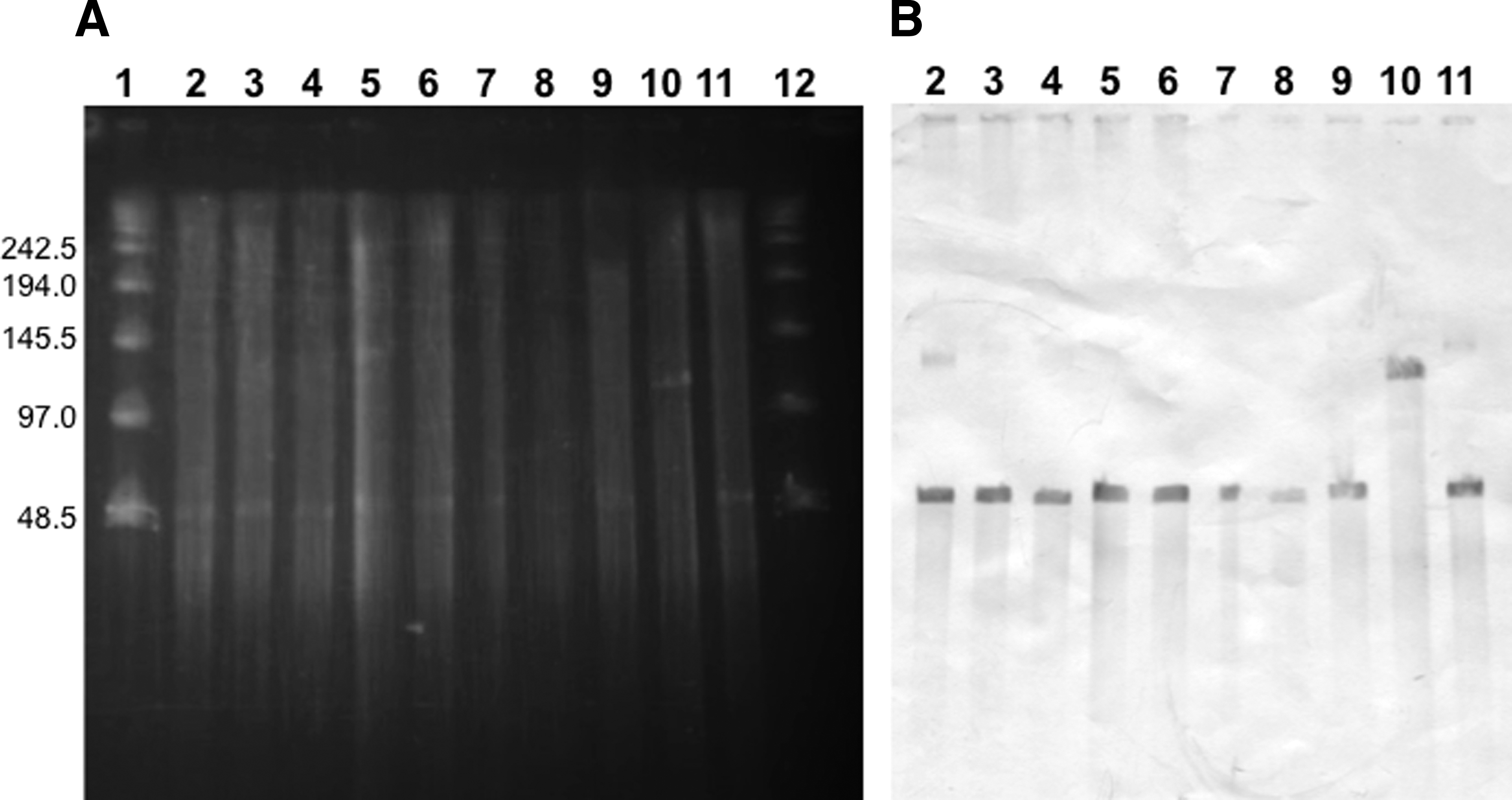
Plasmid profiles of CIP-resistant Escherichia coli transformants.
TSI, triple sugar iron agar slants.
K/A, K over A, refers to an alkaline slant and acid butt.
Relationship between quinolone resistance determinants and level of quinolone resistance
The relationship between quinolone resistance determinants and the MICs of NAL, NOR, and CIP for each isolate are shown in Table 3. Among the 138 gyrA mutants, all but two isolates showed high-level resistance to NAL (MICs ≥256 mg/L). The double mutant with Ser83Phe/Asp87Tyr showed a fully resistant phenotype to NAL (≥ 256 mg/L), NOR (16 mg/L), and CIP (4 mg/L). In contrast, single codon mutants were found to be associated with a wide range of MICs for NOR (0.19–4 mg/L) and CIP (0.032–2 mg/L).
Isolates harboring qnrS1 and wild-type gyrA exhibited an atypical quinolone resistance phenotype that could be identified as susceptible or low-level resistance to NAL (MIC ≤32 mg/L) and decreased susceptibility to CIP (≥ 0.125 mg/L). 26 One qnrS1-carrying isolate with an Ser83Tyr substitution in gyrA presented high MICs for all quinolones: NAL (≥256 mg/L), NOR (3 mg/L), and CIP (2 mg/L). Two isolates did not have gyrA, gyrB, or parC QRDR mutations or qnr genes, although they showed high resistance to NAL (≥256 mg/L) and decreased susceptibility to CIP.
Relationship between quinolone resistance determinants and MLVA types
Asp87Tyr and Ser83Tyr, two dominant mutants observed in the isolates, showed an opposite trend of prevalence. Indeed, the frequency of Asp87Tyr significantly increased from 38.9% in 2004 to 74.4% in 2007 (p = 0.003), while that of Ser83Tyr significantly decreased from 52.8% to 11.6% during the same period (p = 0.001). The mutants were identified at similar frequencies in both blood and stool specimens (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/mdr).
MLVA types of isolates were analyzed and are shown with the number of repeat units in the order of VNTR loci SE1-SE2-SE5-SE7-SE9 (Supplementary Table S2). Asp87Tyr was strongly associated with type 5-5-11-7-3, while Ser83Tyr was related to another type 5-5-9-7-3. Most qnrS1-carrying strains were associated with type 5-5-11-7-3 and resistant to both ampicillin (AMP) and streptomycin (STR) (Table 4). All eight isolates harboring Ser83Ile were found to have similar MLVA types with no amplification of locus SE7 (Table 6). All the resistance determinant types and MLVA types were widely distributed in Thailand (Supplementary Table S3).
CTX, cefotaxime; SXT, trimethoprim/sulfamethoxazole.
Discussion
To elucidate the quinolone resistance acquisition mechanism, 190 S. Enteritidis clinical isolates were investigated, 158 were elucidated to be intermediately or fully quinolone resistant, and 156 were identified to carry quinolone resistance determinants examined in this study. Among these, 138 isolates had mutations in gyrA and 19 isolates carried qnrS1. The MIC50, at which 50% of isolates (i.e., 50% of the population) are inhibited, for NAL was ≥256 mg/L, while that for NOR and CIP was 0.75 and 0.125 mg/L, respectively (Table 3). MIC50s of NAL, NOR, and CIP for isolates with qnrS1 were 24, 1.5, and 0.38 mg/L, respectively.
In contrast, those for isolates with GyrA amino acid substitutions were
In contrast, all isolates with qnrS1 and all isolates with GyrA single amino acid substitutions were susceptible to NOR. Similarly, all isolates with qnrS1 and 137 isolates with single GyrA single amino acid substitution were susceptible to CIP. An isolate with both qnrS1 and GyrA amino acid substitution (Ser83Tyr) was intermediately resistant to CIP. Only one isolate with GyrA double amino acid substitution (Ser83Phe + Asp87Tyr) was resistant to both NOR and CIP. These results confirmed the higher potency of fluorinated quinolones as DNA gyrase inhibitors against strains with qnrS1 and GyrA single amino acid substitutions or both.
In the current study, gyrA mutations causing single amino acid substitutions associated with a higher MIC of NAL and lower susceptibility to CIP were observed, which concurs with data from studies of clinical S. Enteritidis isolates in other countries.7,27,28 In addition, the highest MICs of CIP and NOR were estimated for an isolate with a double mutation causing two amino acid substitutions. This finding is in agreement with previous reports describing the Salmonella requirement of multiple mutations for high-level resistance to fluoroquinolones.14,29
In our study, the mutation causing an amino acid substitution at codon 87 was predominantly Asp87Tyr (83/138), which was also found in many other countries, including Spain, 28 the United Kingdom, 8 Norway, 30 and Ireland. 31 Ser83Tyr was the second predominant mutation (40/138) and was also observed in Norway. 30 In our study, we found strong correlations between specific gyrA mutations and MLVA types (Supplementary Table S2). Interestingly, unlike the cases in Norway, which were associated with travel to Southeast Asia, our data suggest a clonal expansion of quinolone-resistant strains within Thailand.
We identified Ser83Ile in eight isolates distributed nationwide in Thailand (Table 6). This is the second finding all over the world following that reported from Malaysia. 32 All isolates with this substitution presented high MICs of NAL (≥ 256 mg/L), NOR (0.25–1 mg/L), and CIP (0.094–0.38 mg/L) (Table 3). In addition, isolates with Ser83Ile possessed similar MLVA types, of which different alleles were observed only at a single locus in two isolates (Table 6). Interestingly, all eight isolates with Ser83Ile harbored an identical null allele in locus SE7, which is in the clustered regularly interspaced short palindromic repeats, which are known to be highly polymorphic in Salmonella. 26
This evidence indicates that strains with this new mutation might have originated from a single source that clonally expanded throughout Thailand under certain selective pressures. At codon 83, serine (T
In our current study, we found that 19 of 190 S. Enteritidis isolates (10%) carried qnrS1. In contrast, S. Enteritidis strains carrying the qnrS1 gene are rarely reported. For example, a study in 13 European countries found 125 Salmonella isolates carrying qnrS1, but only two were S. Enteritidis from pigs in Poland. 23 Limited number of S. Enteritidis isolates from neonatal calf diarrhea in Egypt carried qnrS. 33
In Asian countries, qnrS was identified in several clinical Salmonella serovars mainly from serovar other than Enteritidis as reported in Korea 34 and Taiwan. 15 The report from China also reported low percentage of qnrS in S. Enteritidis (0.8%, 1/126). 35 In Thailand, qnrS1 was previously detected in 16 S. Corvallis isolated from humans and food animals 13 and recently observed in clinical S. Stanley and S. Anatum from poultry. 36 To our best knowledge, this is the first work reporting clinical S. Enteritidis carrying qnrS1 in Thailand.
As qnrS1 is a PMQR gene, qnrS1 in S. Enteritidis may have resulted from the acquisition of transferable plasmids from other bacteria. The existence of transferable plasmids carrying qnrS1 with the size of 104.7 kbp and 52.7 kbp in strain ID134 and others, respectively, was confirmed by the nuclease S1 analysis of CIP-resistant E. coli transformants (Fig. 2). The transferability of the plasmid carrying qnrS1 was also confirmed by conjugation (Table 5). The AMP-resistant phenotype of transconjugants (Table 5) supported the idea of the coexistence of qnrS1 and the gene encoding β-lactamase, bla, in the same plasmid as has been reported in Klebsiella pneumoniae (blaSHV-2) 37 and S. enterica serovar Stanley (blaLAP-2). 38 This will be elucidated by our further study.
We found that all qnrS1-carrying strains with wild-type gyrA also showed atypical quinolone-resistant phenotype with decreased susceptibility to CIP and susceptible or low-level resistance to NAL (CIPDS NALS/LR; Table 4), which is consistent with the observations reported by Gunell et al. 39 In contrast, other studies demonstrated that qnrS in Salmonella caused other resistance phenotypes, including susceptibility to both CIP and NAL (CIPS NALS) 28 and decreased susceptibility to CIP and resistance to NAL (CIPDS NALR). 33
In the present study, we also found an isolate carrying qnrS1 with a gyrA mutation that showed high MICs for NAL (≥ 256 mg/L) and CIP (2 mg/L), which is consistent with observations in Korea 40 and the United Kingdom. 14 These findings seemed to suggest that qnrS could enhance CIP resistance through the gyrA mutation. In fact, phenotype differences in isolates with qnrS may be associated with the level of Qnr protein expression. Similar to a single mutation in gyrA, carriage of qnrS alone does not confer strong fluoroquinolone resistance to S. enterica. Thus, reducing the quinolone pressure (e.g., NAL) on resistant strains could be crucial to prevent the emergence and spread of S. Enteritidis resistant to fluoroquinolone (e.g., CIP).
Since we found two S. Enteritidis isolates that were resistant to NAL and reduced susceptibility to CIP without qnrA, B, and S1 genes and target mutations in type IV topoisomerase-encoding genes, the other mechanisms such as an alteration of permeability, 35 an enzymatic inactivation encoded by aac(6′)Ib cr, and effux pump encoded by qep and oqxAB10,11,41 might be playing roles in developing resistance to quinolones. Further investigation of other PMQRs will be conducted by our future study.
In conclusion, quinolone resistance of S. Enteritidis in Thailand was found to be mediated predominantly by gyrA mutations that caused amino acid substitutions Asp87Tyr and Ser83Tyr as well as a new amino acid substitution, Ser83Ile. A strong correlation between mutations and specific MLVA types suggested a possible clonal expansion nationwide in Thailand. In addition, the presence of a plasmid-mediated quinolone-resistant gene, qnrS1, raises concerns about a broad dissemination of resistant strains. Based on our data, it is recommended to restrict the usage of quinolone for therapeutic and farming purposes to effectively control the emergence and spread of fluoroquinolone-resistant S. Enteritidis.
Footnotes
Acknowledgments
This work was supported, in part, by a Department of Microbiology Grant, the China Medical Board, the Faculty of Public Health of Mahidol University, and the National Science and Technology Development Agency, Ministry of Science and Technology, Bangkok, Thailand, to O.S.; in part, by a grant from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan, for the Joint Research Program of the Research Center for Zoonosis Control, Hokkaido University to Y.S.; and, in part, by J-GRID and the Japan Initiative for Global Research Network on Infectious Diseases from MEXT to Y.S.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.