
Abstract
GpsB, a key regulator of cell division in Gram-positive bacteria, interacts with a key peptidoglycan synthase at the cell division septum, the penicillin binding protein PBP1 (a.k.a. PonA). Bacillus subtilis GpsB has been reported to interact with other components of the cell division machinery, including EzrA, MreC, and PrkC. In this study, we report an analysis of the arrangement of subunits in Listeria monocytogenes GpsB by small-angle X-ray scattering. The resulting model has an elongated shape with residues critical for interaction with PBP1 and the cell membrane clustered at one end of the molecule. Mutations that destabilize the hexameric assembly of the wild-type protein have a gpsB null phenotype, indicating that oligomerization is critical for the correct function of GpsB. We suggest a model in which a single GpsB hexamer can interact with multiple PBP1 molecules and can therefore influence the arrangement of PBP1 molecules within the cell division machinery, a dynamic multiprotein complex called the divisome, consistent with a role for GpsB in modulating the synthesis of the cell wall.
Introduction
B
The importance of GpsB in mediating the coordination of PBP activity with other divisome components is underlined by the interaction network in which GpsB participates. First, GpsB has been reported to interact with EzrA in two hybrid assays, although only unidirectionally, 5 potentially providing an indirect link between FtsZ and PBPs, given that numerous in vitro and in vivo studies have shown that EzrA interacts with FtsZ (e.g., 6–9). Second, it has been reported that GpsB is phosphorylated in B. subtilis by PrkC, 19 a eukaryotic-like membrane-embedded Ser/Thr kinase with an extracellular PASTA domain 20 that binds peptidoglycan fragments. 21 The relationship between GpsB and PrkC suggests that GpsB could be involved in transducing alterations in cell wall structure on the outside of the cell to the interior. Finally, a major portion of the GpsB sequence is homologous to the lipid binding domain of the DivIVA protein; the lipid binding domain promotes the localization of DivIVA to negatively curved lipid membranes, 22 which is pivotal to DivIVA's role in promoting the assembly of the divisome at midcell. 23 Whether GpsB regulates cell wall synthesis by similarly localizing to curved membranes remains to be clarified.
To help elucidate further the functional role of GpsB, we recently solved crystal structures of its component domains from L. monocytogenes (LmGpsB) and B. subtilis (BsGpsB). 11 GpsB proteins are typically approximately 100 amino acids in length and contain two domains, with the 65–70 amino acid N-terminal domain (N-GpsB) resembling the lipid binding domain of DivIVA, a parallel coiled-coil dimer. 23 The 20–25 amino acid C-terminal domain (C-GpsB) forms a parallel triple helical coiled-coil. 11 While the N- and C-terminal domains in isolation assemble into dimers and trimers, respectively, the full-length GpsB protein is a hexamer, 11 which to date has been recalcitrant to successful crystallization. In this study, we present a study of the arrangement of subunits in LmGpsB using small-angle X-ray scattering (SAXS) and a mutational analysis of candidate oligomer stabilizing residues. To understand the role of GpsB in cell division, we have also analyzed the enzymatic properties of PBP1 in the presence and absence of BsGpsB. In combination, these data are consistent with a model where GpsB serves to localize multiple copies of PBP1, a process that appears to be critical for efficient cell division.
Materials and Methods
Bacterial strains and growth conditions
All bacterial strains used in this study are listed in Table 1. L. monocytogenes strains were grown in brain–heart infusion (BHI) broth or on BHI agar plates at 37°C. When necessary, antibiotics and supplements were added to the growth media at concentrations of 50 μg/ml for kanamycin and 1 mM for IPTG. The Escherichia coli strain TOP10 was used for all cloning procedures. 24
The arrow (→) stands for a transformation event.
Construction of plasmids and strains
The T88A, T88D, F91A, L94A, and F105A amino acid exchanges were introduced into L. monocytogenes gpsB, cloned previously into pSH254, by Quikchange mutagenesis 25 using the primer pairs JR290/JR291, JR291/JR293, JR311/JR312, JR313/JR314, and JR315/JR316, respectively (Table 2). The presence of the desired mutations was verified by DNA sequencing. The resulting plasmids are listed in Table 1 and were introduced into L. monocytogenes EGD-e by electroporation. 26 Kanamycin-resistant clones were selected, and plasmid insertion at the attB site of the tRNAArg locus was verified by PCR.
To generate the plasmid for expressing C-LmGpsB, the open reading frame encoding L. monocytogenes GpsB residues 87–113 was PCR amplified from genomic DNA from strain EGD-e with primers RC87f and RC113f, which contain flanking NcoI and XhoI restriction sites, and cloned between the NcoI and XhoI sites of pMAT11, a modified form of pHAT4. 27 The plasmids for expressing the full-length LmGpsBF91A, LmGpsBF105A mutants and the double mutant LmGpsBF91AF105A were prepared by Quikchange mutagenesis using primers RC91f, RC91r, RC105f, and RC105r; for the double mutant, two rounds of mutagenesis were used—the first round with primer pairs RC91f and RC91r and the second round with primer pairs RC105f and RC105r (Table 2).
Similarly, the full-length BsGpsBT75E and BsGpsBT75D mutants were prepared by Quikchange mutagenesis with primers RC75Ef and RC75Er, and RC75Df and RC75Dr, respectively; the plasmid for expressing full-length wild-type BsGpsB was used as a template for the Quikchange reaction to generate the T75D mutation. By contrast, for the T75E Quikchange reaction, the plasmid for expressing BsGpsBT75D was used as a template. The resultant plasmids were sequenced to verify the successful introduction of the desired mutations and were subsequently used for the overproduction of the full-length GpsB variants by the same procedures as wild-type GpsB proteins. 11
Isolation of cellular proteins and Western blotting
Cells were harvested by brief centrifugation in a table-top microfuge and washed with ZAP buffer (10 mM Tris-HCl pH 7.5, 200 mM NaCl). Cells were resuspended in 1 ml ZAP buffer containing 1 mM PMSF and disrupted by sonication. The cell debris was removed by centrifugation, and the supernatant was used as the soluble protein extract. Protein samples were separated by SDS polyacrylamide gel electrophoresis and then transferred onto positively charged polyvinylidene fluoride (PVDF) membranes using a semidry transfer unit. GpsB and DivIVA were detected using polyclonal rabbit antisera raised against L. monocytogenes GpsB 11 and B. subtilis DivIVA, 28 respectively, with anti-rabbit immunoglobulin G conjugated to horseradish peroxidase as the secondary antibody. The ECL chemiluminescence detection system (Thermo Scientific) was then used for detection of the peroxidase conjugates on the PVDF membranes in a chemiluminescence imager (Vilber Lourmat).
Protein purification
PBP1 and GpsB proteins were purified as described previously. 11 C-LmGpsB proteins were expressed as fusion proteins with N-terminal His6- and MBP-tags; the protocols for expression and purification of the fusion protein by Ni-NTA chromatography were as described previously for C-BsGpsB. 11 Following Ni-NTA chromatography, the fusion protein was cleaved with TEV protease (ratio of fusion protein:TEV 50:1 by mass) overnight at 4°C while simultaneously dialyzing into a buffer of 50 mM Tris-HCl pH 8.0, 300 mM NaCl using a 2 kDa molecular weight cutoff dialysis membrane. The dialysate was passed back over a 5 ml Ni-NTA cartridge (Qiagen) to separate TEV protease, C-LmGpsB released by cleavage from the fusion protein and residual uncleaved His6-MBP-C-LmGpsB. The flow through from the Ni-NTA column was concentrated in a centrifugal concentrator to 1 ml and loaded onto a Superdex75 XK16/60 column (GE Healthcare) equilibrated in a buffer of 10 mM Tris-HCl pH 8.0, 250 mM NaCl. Elution fractions containing C-LmGpsB were pooled, concentrated to c. 5 mg/ml in a centrifugal concentrator, and then dialyzed further into 20 mM sodium phosphate pH 7.8, 250 mM NaCl before finally flash freezing the protein in liquid nitrogen and storing at −80°C.
Size exclusion chromatography
The full-length wild-type LmGpsB and LmGpsBF91A, LmGpsBF105A and LmGpsBF91AF105A proteins were analyzed on a Superdex200 Increase 10/300 GL column (GE Healthcare) equilibrated in 10 mM Tris-HCl pH 8.0, 250 mM NaCl, at a flow rate of 0.5 ml/min. Protein samples at 3 mg/ml concentration were injected onto the column through a 100 μl sample loop. Wild-type BsGpsB and the BsGpsBT75D and BsGpsBT75E mutants were analyzed under the same conditions, except the protein concentration was 1 mg/ml.
Size exclusion chromatography-MALS
GpsB samples (500 μl) at concentrations of 8 mg/ml or 0.5 mg/ml were loaded onto a Superdex200 Increase 10/300 GL column (GE Healthcare) equipped with a Jasco UV-2077 detector, Wyatt DAWN Heleos II EOS 18-angle laser photometer (with the 13th detector replaced with the QELS in-line dynamic light scattering detector) coupled to a Wyatt Optilab rEX refractive index detector. The flow rate was 0.75 ml/min. Molecular mass and concentrations of the peaks eluting from the column in a running buffer of 10 mM Tris-HCl pH 8.0, 250 mM NaCl were analyzed using Astra 6.2 (www.wyatt.com/products/software/astra.html).
Circular dichroism
CD thermal melts were measured in a 1-mm path length quartz cuvette in a 20 mM sodium phosphate pH 7.8, 200 mM NaCl buffer for wild-type LmGpsB, N-LmGpsB, and C-LmGpsB and in 20 mM sodium phosphate pH 7.8, 250 mM NaCl for wild-type BsGpsB, BsGpsBT75D, and BsGpsBT75E. The temperature was increased at a rate of 1°C/min, and ellipticity was monitored at a wavelength of 222 nm with a response time of 8 seconds and bandwidth of 2 nm. The reversibility of the melts was verified by recording spectra in the wavelength range 200–240 nm (scan speed 20 nm/min, response 2 seconds) before and after the melt, to check that the spectra were superimposable. Protein concentrations were 23 μM for wild-type LmGpsB, N-LmGpsB, and C-LmGpsB, 29 μM for LmGpsBF91AF105, and 5 μM for wild-type BsGpsB, BsGpsBT75D, and BsGpsBT75E.
The CD spectra of wild-type and C-LmGpsBF105A proteins were recorded at protein concentrations of 23 and 33 μM, respectively, in a buffer of 20 mM sodium phosphate pH 7.8 at 20°C. The scan speed was 10 nm/min, and the response time was 4 seconds. Protein concentrations were determined based on calculated extinction coefficients at 280 nm 29 for LmGpsB, N-LmGpsB, and BsGpsB proteins; for the C-LmGpsB proteins, which lack tryptophan and tyrosine residues, amino acid analysis was used instead to quantify concentration. Secondary structure composition was evaluated from the spectra using the program CDSSTR 30 within the DICHROWEB 31 server.
SAXS data collection and processing
The LmGpsB and N-LmGpsB proteins were analyzed in a buffer of 10 mM Tris-HCl pH 8.0, 150 mM NaCl at a protein concentration of 40 mg/ml. SAXS intensity data, I(q) versus q, (
For SEC-SAXS data collection at Diamond, 50 μl of LmGpsB was loaded onto a Superdex 200 Increase 3.2/300 column and the eluent flowed through the SAXS beam at a flow rate of 0.1 ml/min; the buffer used as the blank in these measurements was the eluent after one SEC column volume. SAXS data were constantly collected at 1 second intervals using a 2 M Pilatus detector (Dectris) at a distance of 3.9 m and an X-ray wavelength of 1 Å. Subtraction of the SEC-SAXS data was completed for each frame across the elution peak, and the radius of gyration (Rg) and the integral of ratio to background were plotted. The data were scaled, merged, and averaged for each frame with a consistently similar Rg. All further processing and analysis of data were carried out using ScÅtter (www.bioisis.net/scatter).
Pair distance distribution functions were calculated with GNOM, 34 with Dmax values for LmGpsB and N-LmGpsB of 185 and 76 Å; the Dmax values were estimated initially using DATGNOM and the atomic coordinates of N-LmGpsB, 11 from which the pair distance distribution function was calculated with ScÅtter; all waters and heteroatoms were removed before the calculation.
SAXS model generation
Dummy atom models were constructed in DAMMIN in slow mode using scattering data up to a maximum q value of 8/Rg (0.15Å−1 for LmGpsB, 0.36Å−1 for N-LmGpsB). For N-LmGpsB, the chi-squared values calculated by DAMMIN, corresponding to the agreement between experimental scattering curves and scattering curves calculated from the dummy atom models, ranged between 1.60 and 1.64. For LmGpsB, the chi-squared values from individual DAMMIN run were in the range 1.11–1.14. For N- LmGpsB, the average normalized structural discrepancy (NSD) between models from 10 independent DAMMIN runs, after averaging them together in the DAMAVER suite, 35 was 0.539 ± 0.09. For LmGpsB, the models from 15 DAMMIN runs were superimposed and averaged in the DAMAVER suite and the averaged model was then input into DAMSTART to generate a starting model for a further 15 DAMMIN runs. The final models were then again averaged together using the DAMAVER suite; the NSD between models was 0.552 ± 0.09.
Dummy atom models were visualized in CHIMERA 36 by representing the atoms as beads, with the bead scale adjusted to a value equivalent to the dummy atom radius divided by 0.7. The surface mesh representation of dummy atom models was generated by using the CHIMERA “molmap” command to convert the model to the equivalent electron density map at 25 Å resolution—the contouring of this map was then adjusted to enclose the surface of the dummy atoms. The crystal structures of the N- and C-terminal domains of GpsB 11 PDBids 4ug1 and 5an5, respectively, were also represented as 25 Å resolution electron density maps using the “molmap” command in CHIMERA. For the LmGpsB sample, the molecular weight of the scattering particles was calculated from the volume of correlation 37 using ScÅtter.
Production of lipid II-meso-diaminopimelic acid
Lipid II-meso-diaminopimelic acid (Lipid II-m-DAP) was synthesized using UDP-MurNAc-pentapeptide isolated from Bacillus cereus, essentially as described previously38,39 with the following modifications. Purification was performed over a DEAE-cellulose column using a linear gradient of chloroform/methanol/water (2:3:1 v/v/v) to chloroform/methanol/1 M ammonium bicarbonate (2:3:1 v/v/v). The fractions containing Lipid II-m-DAP were collected and dried under vacuum. The resulting lipid II-m-DAP was then dissolved in 1:1 chloroform:methanol and stored at −20°C until use.
Production of [ 14 C]-amidated Lipid II-m-DAP
The ability of MurG to exchange the GlcNAc group between UDP-GlcNAc and Lipid II was used to radioactively label Lipid II. Since the E. coli MurG used here displayed much lower affinity for amidated Lipid II-m-DAP than the nonamidated form, we first synthesized [ 14 C]-Lipid II-m-DAP and then amidated this using AsnB from B. subtilis. First, 1.22 μmol of purified lipid II-m-DAP was incubated with 12.5 μCi of UDP-N-acetyl-D-[1- 14 C]glucosamine (specific activity 55 mCi/mmol) (Hartmann Analytic GmbH). The labeling reaction was performed in 1 ml of 100 mM Tris-HCl, pH 8.0, 1 mM MgCl2, and 2% (w/v) Triton X-100. The reaction was started by the addition of 1 μL purified recombinant E. coli MurG, which is able to exchange the N-acetyl-D-glucosamine of lipid II with that of UDP-N-acetyl-D-[1- 14 C]glucosamine. After incubation at room temperature for 2.5 hours, the reaction was complete as determined by liquid scintillation counting.
To amidate the carboxylic acid group of the m-DAP, lipid II-DAP was incubated with B. subtilis AsnB in the presence of ATP and glutamine (to be published elsewhere), followed by extraction using butanol/pyridine acetate, pH 4.2, and another purification step over a DEAE cellulose column using a gradient of chloroform/methanol/water (2:3:1 v/v/v) to chloroform/methanol/0.5 M ammonium bicarbonate (2:3:1 v/v/v).
In vitro peptidoglycan synthesis assays
The continuous fluorescence glycosyltransferase (GTase) assay using dansyl-labeled lipid II as the substrate was performed essentially as described previously, 40 with only minor modifications to buffer conditions and temperature. We also used an endpoint assay to measure the GTase and transpeptidase (TPase) activity under the same experimental conditions. The activity of BsPBP1 (0.4 μM) was measured at 37°C in the absence and presence of 10 μM BsGpsB proteins in 50 mM HEPES.NaOH pH 7.5, 20 mM NaCl, 10 mM CaCl2, and 5% (v/v) glycerol. The endpoint assay used [ 14 C]-labeled native amidated lipid II-m-DAP substrate followed by the quantification of both GTase and TPase products by high-pressure liquid chromatography (HPLC) as described previously. 41
Results
Low resolution model of subunit arrangement in solution by SAXS
In the absence of diffracting crystals of full-length LmGpsB, SAXS was used to determine its low resolution structure since the correct assembly of subunits in LmGpsB is highly pertinent to the protein's function. To validate this approach, we first analyzed N-LmGpsB for which the crystal structure has already been solved. 11 The measured scattering profile for N-LmGpsB matched favorably with the theoretical profile calculated from the corresponding atomic coordinates (Fig. 1A). From a Guinier analysis of the scattering profile, an Rg of 21.4 Å was obtained, while the pair distribution function calculated by inverse Fourier transformation with GNOM 34 gave an Rg of 22.2 Å. These values compare extremely favorably with an Rg value of 21.4 Å that was calculated from the atomic coordinates with CRYSOL. 42 Finally, ab initio dummy atom molecular models built from the scattering data with DAMMIN 43 reproduced well the shape and dimensions of the dimeric N-LmGpsB (Fig. 1B).
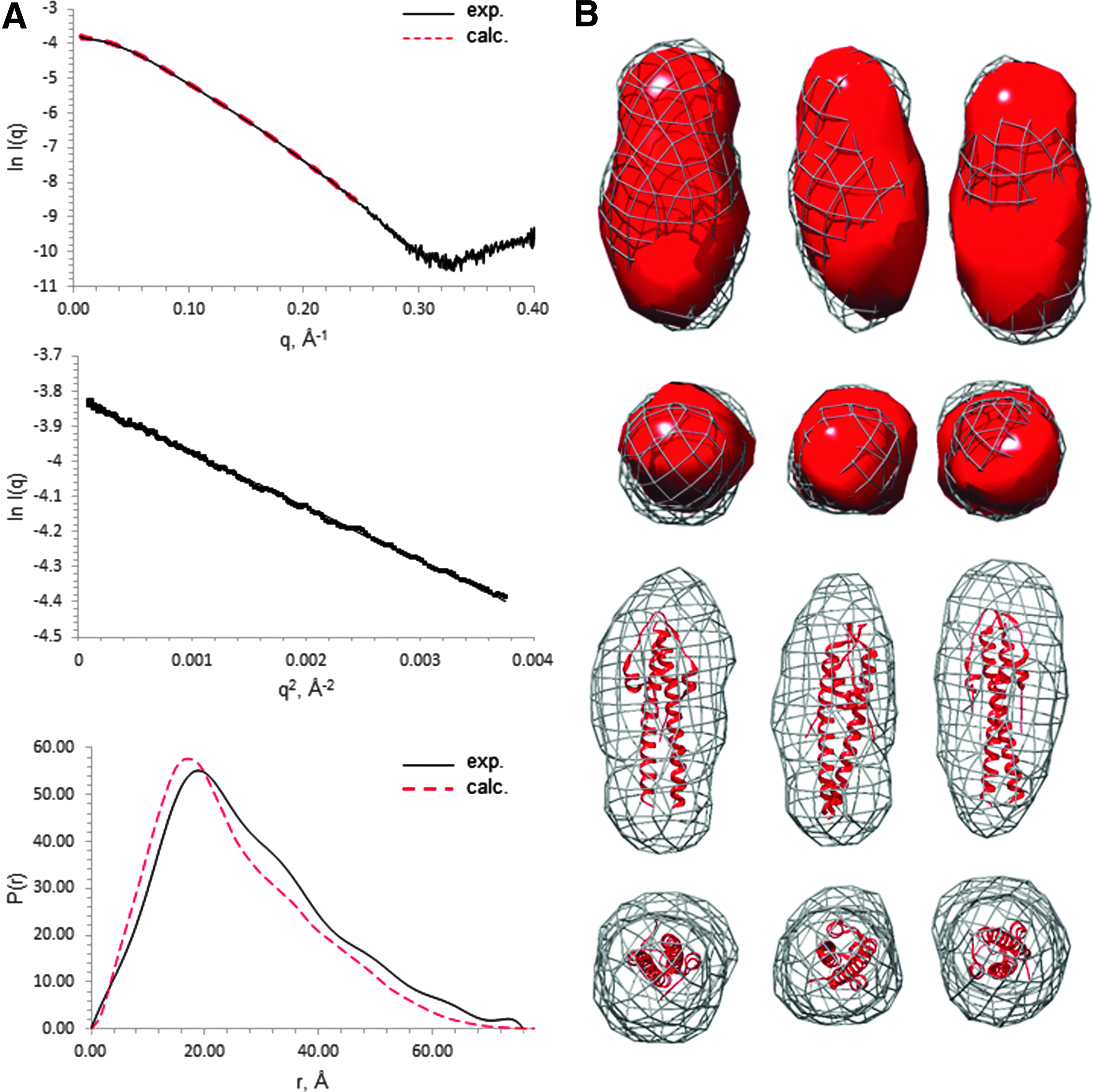
SAXS analysis of N-LmGpsB.
Analysis of LmGpsB yielded the best quality data on SAXS beamlines with an in-line SEC facility. The monodispersity of LmGpsB samples after in-line SEC was indicated by the linearity of Guinier plots at q values less than 1.3/Rg (Fig. 2A). The molecular mass of the scattering particles after in-line SEC, calculated from the volume of correlation, 37 was 75,900 Da, which is within 5% of the actual mass of the expressed LmGpsB protein when assembled as a hexamer.

SAXS analysis of LmGpsB.
A Porod–Debye analysis of the comparative flexibility of LmGpsB and N-LmGpsB suggested that the full-length protein is more flexible than the isolated N-terminal domain. The Porod–Debye plot for N-LmGpsB rose to a plateau (Fig. 2B), which is characteristic of a folded compact protein. 44 By contrast, the Porod–Debye plot for LmGpsB had a less pronounced plateau and instead conformed to a hyperbole. The relatively shallow gradient of the asymptote, however, indicated that LmGpsB is markedly less flexible than a completely unfolded protein. Given the successful crystal structure determinations of both N- and C-terminal domains of GpsB, the flexibility within LmGpsB is most likely attributable to the linker region between the domains.
The pair distribution function of LmGpsB had, like N-LmGpsB, the characteristics of an elongated molecule, 45 with a steep initial increase in P(r) to a maxima followed by a more gradual decrease to zero at Dmax. From the Guinier plot and pair distribution functions, Rg values of 51.2 and 53.1 Å were obtained for LmGpsB. Ab initio dummy atom molecular models were calculated in DAMMIN without symmetry constraints, however, no obvious symmetric relationships were apparent from the resultant models. A notable feature of the models is two distinct lobes of different sizes connected by a narrower central region (Fig. 3).
SAXS molecular envelopes for LmGpsB.
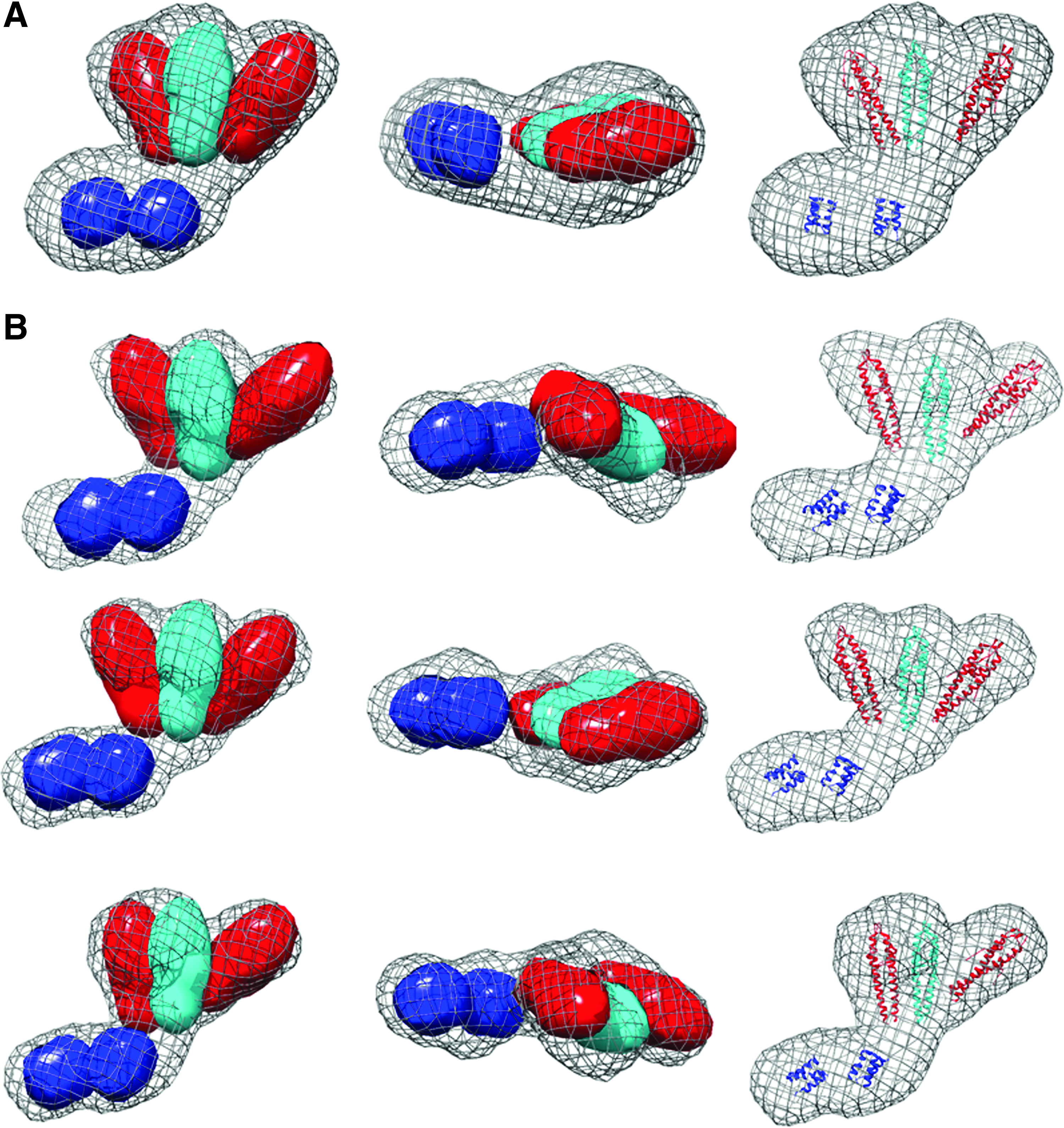
The most logical arrangement of the individual domains in the GpsB hexamer resembles a tripod with the three dimeric N-GpsB domains aligned as in the legs in a tripod with the two C-GpsB trimeric domains encompassing its base (Fig. 4A). Such an arrangement is logical on the basis that it presents a relatively straight path and near equivalent distance between connected C- and N-terminal domains of GpsB. Manually positioning the atomic coordinates of the N- and C-terminal domains of GpsB into the ab initio dummy atom model supports such a tripod arrangement (Fig. 3); the three N-GpsB dimers encompass the larger of the two lobes, and the two C-GpsB trimers correspond to the smaller one.
Interaction between GpsB domains.
The importance of hexamer formation for GpsB activity in vivo
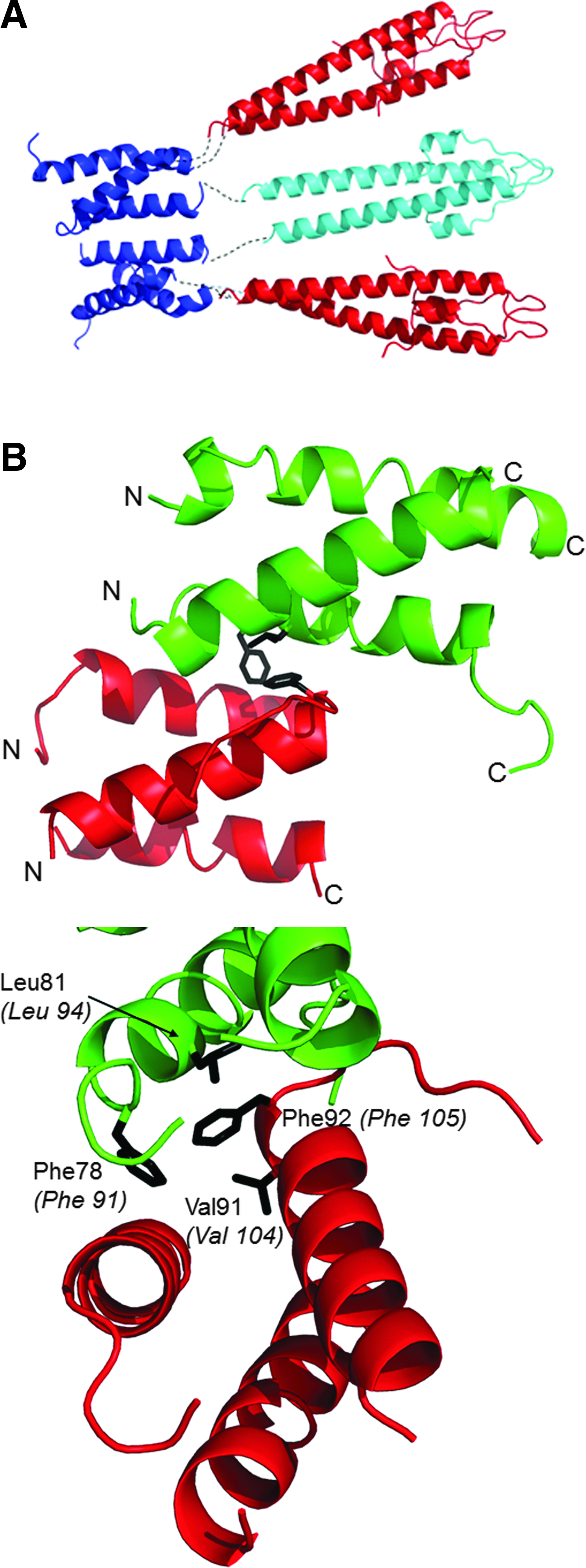
The tripod model positions the two C-LmGpsB trimers in close proximity. The lattice interactions in the crystal structure of C-BsGpsB (encompassing residues 76–98 of BsGpsB) are thus of interest in considering how the subunits assemble to form the GpsB hexamer. Two highly conserved phenylalanines (F78, F92) stand out for being highly solvent exposed on the surface of the C-BsGpsB trimer; within the crystal lattice both F78 and F92 are buried at the interface between adjacent trimers (Fig. 4B). To probe whether burial of these residues drives the assembly of subunits within the full-length hexameric GpsB, the equivalent residues in LmGpsB, F91, and F105 were mutated to alanine, both singly and in combination. The oligomeric state of the wild-type and the mutant proteins was analyzed by SEC. Mutation of F105 to alanine both alone and in combination with the F91A mutation substantially increased the retention volume of LmGpsB mutants on SEC analysis relative to the wild-type LmGpsB (Fig. 5A). Subsequent SEC-MALS analysis of the LmGpsBF91AF105A double mutant confirmed this is due to a change in the oligomeric state; the double mutant formed a trimer at high protein concentrations and a dimer at lower concentrations (Fig. 5B). Mutation of F91A alone had an intermediate effect on assembly, with SEC analysis revealing two species; the predominant peak has a similar retention volume as the hexameric wild-type protein, while the other peak has a retention volume more similar to the F91AF105A double mutant (Fig. 5B).
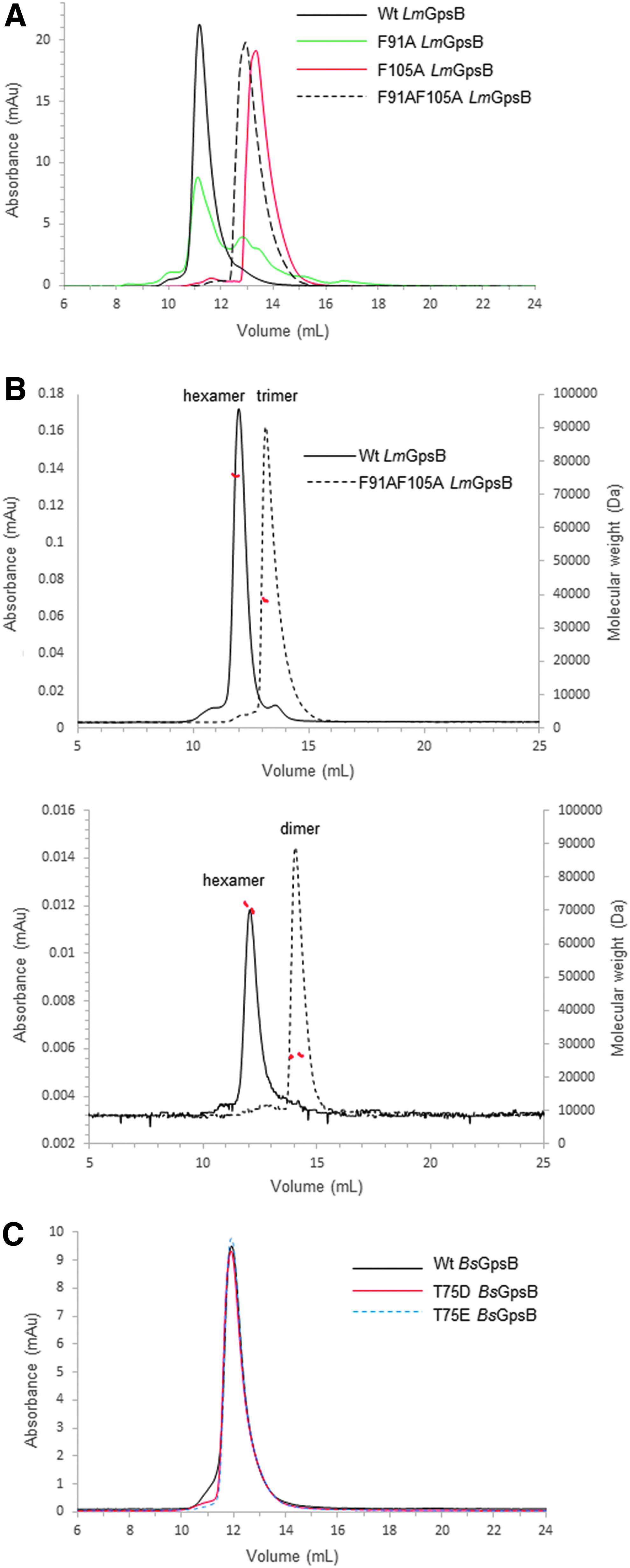
SEC and SEC-MALS analysis of GpsB proteins.
The structural importance of F91 and F105 is further supported by thermal stability analysis using circular dichroism spectroscopy. Wild-type LmGpsB has a Tm (the temperature of the midpoint of thermal unfolding) of 50°C, whereas the LmGpsBF91AF105A double mutant has a Tm of 45°C (Fig. 6A). At the protein concentrations used for the CD analysis, the LmGpsBF91AF105A double mutant was observed to be a dimer on SEC-MALS analysis (Fig. 6B), which may explain why LmGpsBF91AF105A has a similar Tm as the dimeric, isolated N-LmGpsB domain (Fig. 6A).
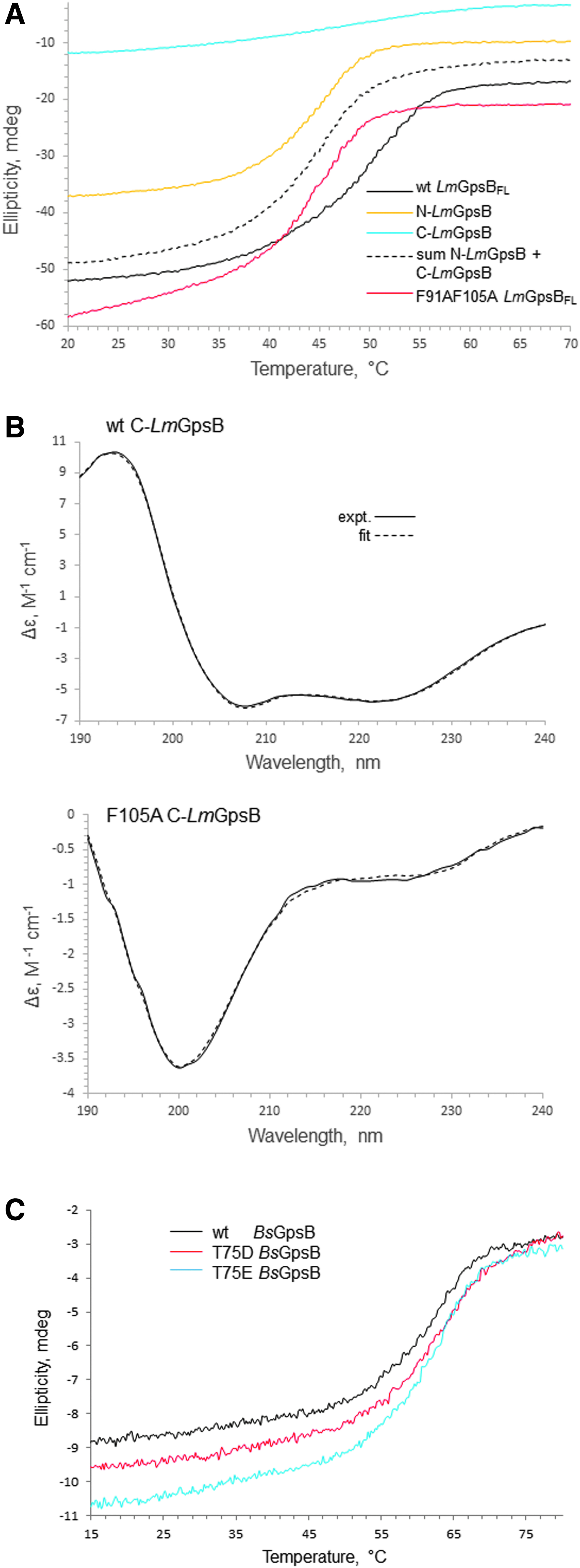
Circular dichroism spectra.
To further investigate the structural role of F105, the alanine substitution was made within the context of the isolated C-terminal domain. The isolated C-terminal domain forms trimers, rather than hexamers, in solution, 11 and F105 is exposed on the surface of this trimer, rather than in the hydrophobic core. F105 is unlikely to have an important structural role within the isolated C-terminal domain, but will stabilize the hexameric full-length protein. However, the C-LmGpsBF105A fold was markedly destabilized as the alpha helical content was reduced from 70% in the wild-type C-LmGpsB protein to 10% in the F105A mutant. This effect can perhaps be reconciled on closer analysis of the structure of C-BsGpsB; F92 (equivalent to F105 in L. monocytogenes GpsB) is in close proximity to the hydrophobic core residue V91, and while one face of the F92 aromatic ring is surface exposed, the other face packs against V91 (Fig. 4B). The structural importance of F105 may therefore have two components; first, F105 stabilizes indirectly the folding of the isolated C-terminal domain because of its interaction with Val104, and second, F105 is likely to stabilize further interactions between C-GpsB trimers within the GpsB hexamer, by packing against Phe91 and Leu94 in another protomer in the hexamer (Fig. 4B). The dimers and trimers observed with the LmGpsBF91AF105A double mutant likely arose from misfolding of at least the C-terminal domain, given the loss of secondary structure associated with the F105A mutation (Fig. 6A).
To validate the effect of mutations in the interface between C-GpsB trimers, we used a complementation assay to measure the effect of gpsB mutations on GpsB activity in L. monocytogenes. Briefly, the L. monocytogenes ΔgpsB mutant grew equally well as the wild-type strain at 30°C and was still viable at 37°C; however, the L. monocytogenes ΔgpsB mutant was not viable at 42°C, neither were gpsB mutants affected in self-interactions or interactions with the L. monocytogenes orthologue of B. subtilis PBP1, PBP A1 (for simplicity referred to herein as PBP1). 11 Mutations that individually exchanged the pair of phenylalanines (F91, F105, the equivalent B. subtilis residues are highlighted on the structure in Figure 4) and the adjacent L94 into alanines were introduced to L. monocytogenes gpsB, and the resulting alleles were tested in the complementation assay. As shown in Fig. 7A, the wild-type L. monocytogenes strain EGD-e proliferated readily at 42°C, whereas the ΔgpsB mutant (strain LMJR19) was unable to grow at all at this temperature. Reintroduction of wild-type gpsB into the ΔgpsB mutant (strain LMS56) repaired this defect, demonstrating successful complementation. The point mutations introduced fell into two classes: strain LMS185, expressing gpsBF91A was able to grow at 42°C, although with increased autolysis in the stationary phase as observed by phase contrast microscopy (not shown) and by growth curve analysis (Fig. 7A), suggesting that the in vivo activity of LmGpsBF91A was partially impaired. By contrast, mutations L94A and F105A completely prevented growth of strains LMS186 and LMS187, respectively (Fig. 7A), indicating that these amino acid exchanges generated biologically inactive GpsB proteins. Western blotting showed that all the GpsB mutant proteins were expressed both during growth at 37°C, where all strains are viable (Fig. 7C), and also 2 hours after a temperature shift to 42°C (Fig. 7D). The loss of growth at 42°C is therefore more likely to result from a loss of a functional property of GpsB rather than a loss of protein production or its degradation.
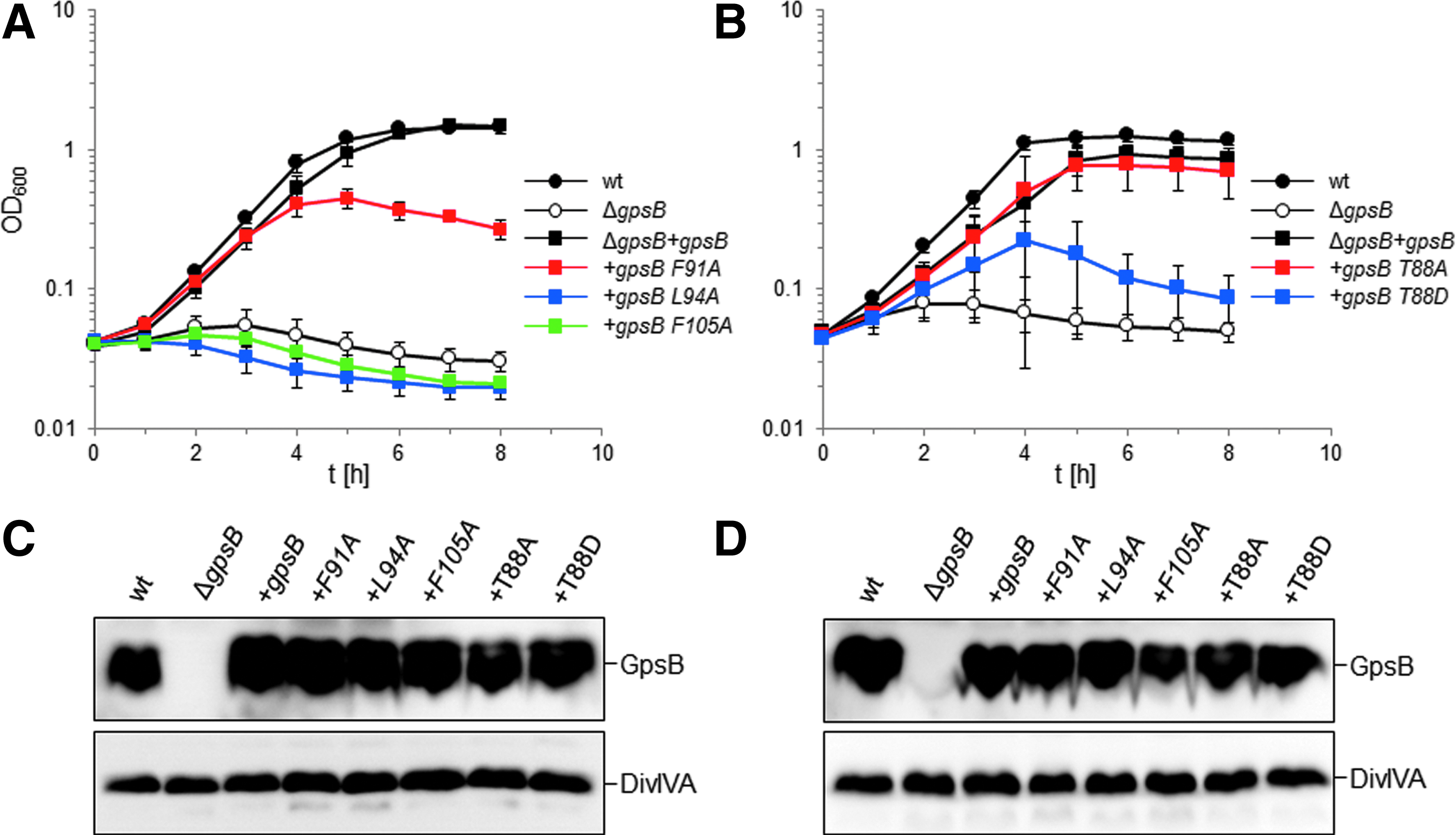
Effect of mutations in the GpsB C-terminus on growth of L. monocytogenes at 42°C.
Role of the phosphorylation of GpsB
It has been reported recently that GpsB was phosphorylated in B. subtilis cells at T75 (T88 in LmGpsB) by the kinase PrkC. 19 The phosphomimetic mutations T75D and T75E appeared to have the same salt-sensitive phenotype as a gpsB deletion. 19 Likewise, we found that the introduction of the analogous T88D exchange into LmGpsB almost completely inactivated the protein. However, the phosphoablative T88A mutation was without effect on complementation activity of LmGpsB (Fig. 7B, D). T75 in BsGpsB and T88 in LmGpsB are located in the linker between the two domains. To investigate whether the T75E and T75D mutations had any impact on the hexamerization of BsGpsB, full-length, wild-type, T75E and T75D mutant BsGpsB proteins (BsGpsBT75E and BsGpsBT75D) were analyzed by SEC in conditions under which wild-type BsGspB was shown by SEC-MALS to form a stable hexamer. 11 The T75E and the T75D mutations did not have a major effect on the size exclusion chromatograms of BsGpsB; the major species in wild-type and both mutant proteins had an identical retention volume (Fig. 5C). Similarly, thermal denaturation analysis by circular dichroism did not reveal any marked difference in the structural stability of wild-type BsGpsB and the T75D and T75E mutants (Fig. 6C).
Effect of BsGpsB on the glycosyltransferase and transpeptidase activity of BsPBP1 in vitro
To explore the functional role of GpsB and the relationship to its structure, the effect of BsGpsB on the GTase and TPase activities of BsPBP1 was investigated. BsPBP1 was used as both the TGase and TPase activities of this enzyme have successfully been recapitulated in vitro, 46 which is not yet the case for the listeria PBPs. Both wild-type and phosphomimetic T75D/T75E mutants of BsGpsB were used in these experiments in case any modulation of PBP1 activity by GpsB requires prior phosphorylation of GpsB. The GTase activity of BsPBP1 was monitored using a fluorescence assay, in which the polymerization of fluorescently labeled lipid II followed by digestion of the resulting glycan chains by a muramidase results in a reduction of fluorescence intensity with time, correlated with the rate of the reaction. The addition of a twenty five-fold excess of either wild-type or the phosphomimetic T75D/T75E mutants of BsGpsB had no apparent effect on the rate of the GTase reaction catalyzed by BsPBP1 (Fig. 8A).

GpsB does not affect the enzyme activities of PBP1 in vitro.
The effect of BsGpsB on BsPBP1 was explored further using a HPLC-based in vitro PG synthesis assay 41 to monitor the composition of peptidoglycan synthesized by BsPBP1 in the presence and absence of BsGpsB. Consistent with others' previous observations, 46 BsPBP1 was active as a TPase when assayed with a native lipid II substrate that had been amidated at the ɛ-carboxylate of meso-diaminopimelic acid (lipid II-m-DAP). In this endpoint assay, PBP1 produced peptidoglycan with approximately 30% of the peptides present in cross-links, irrespective of the absence or the presence of a twenty five-fold molar excess of either wild-type or the phosphomimetic T75D/T75E mutants of BsGpsB (Fig. 8B). As observed with other PBPs, 39 BsPBP1 also exhibited a weak DD-carboxypeptidase activity that was unaffected by the presence of any BsGpsB protein tested. Since BsGpsB interacts with BsPBP1 with a Kd of 0.7 μM, 11 at the protein concentrations used in both assays BsPBP1 should be bound to BsGpsB, suggesting that the interaction has no discernible effect on either the GTase or the TPase activities of BsPBP1 in vitro.
Discussion
The quaternary structure and the connectivity between domains in GpsB are an intriguing geometric puzzle; GpsB is a hexamer, while the isolated N- and C-terminal domains are dimeric and trimeric, respectively. The arrangement of the domains from the SAXS analysis resembles a logical tripod arrangement, but with a more asymmetric shape than perhaps predicted a priori. The asymmetry may be driven, at least in part, by the interactions between C-GpsB trimers; the SAXS envelope suggests a “staggered” alignment of the two C-GpsB trimers, in which one of the trimers is closer to the center of mass of the GpsB hexamer than the other (Fig. 3). A similar staggered alignment is observed in the crystal packing of C-BsGpsB. In this case, each trimeric C-BsGpsB coiled-coil packs in a parallel manner against an adjacent trimer, but offset such that the N-terminal end of one coiled-coil packs against the C-terminal end of the other (Fig. 4B). This arrangement involves hydrophobic interactions between the highly conserved amino acids F78, L81, and F92 in BsGpsB, equivalent to F91, L94, and F105 in LmGpsB. From the SEC analysis, F105 is clearly more important for the stability of the hexamer than F91 (Fig. 5A) and this is fully consistent with the complementation assay, in which mutation of F105, as well as L94, has a larger impact than mutation of F91. The milder effect of the F91A mutation may be attributed to the F105A mutation having a greater impact on the concentration of GpsB hexamers in vivo. Overall, these observations support the conclusion that the interactions between C-GpsB domains in the GpsB hexamer are similar to those observed in the C-BsGpsB crystal lattice, which is not predicted to be stable on analysis of the crystal packing with the PDB-PISA web server. However, weak interactions between isolated C-GpsB domains will be stabilized within GpsB by the covalently attached dimeric N-GpsB domains that will bridge two trimers in a hexamer. Such an effect could explain why the full-length GpsB protein has a higher Tm than the combined CD thermal melts of the isolated domains (Fig. 6A). The stabilizing effect is illustrated schematically in Fig. 4A, in which cyan is used to denote the N-LmGpsB dimer that forms a bridge between two C-LmGpsB trimers within the LmGpsB hexamer. Previous mutagenesis results have demonstrated that the C-terminal domain is essential for formation of GpsB hexamers and that mutations in residues critical for C-GpsB trimer formation render GpsB inactive. 11 It would therefore appear from the loss of GpsB function in strains harboring gpsB alleles that affect GpsB hexamer formation that the correct assembly of GpsB is absolutely critical for its function in vivo.
With the arrangement of domains in the SAXS envelope, the distances between covalently linked domains are not equivalent. This asymmetric arrangement requires the linker between domains to adopt different conformations, and indeed secondary structure prediction analyses predict that the linker is disordered. The flexibility and bendability of the linker can be further reconciled with the high frequency of the disorder promoting residues 47 proline, lysine, methionine serine, and threonine in this region; the 23 amino acid L. monocytogenes GpsB linker has twelve such residues, which are also abundant in the linkers of orthologous GpsB proteins. 11
The position of F91 and L94 close to or at the interface between subunits is particularly interesting in view of the recent identification that, in B. subtilis, the nearby T75 (equivalent to T88 in L. monocytogenes) is phosphorylated by the kinase PrkC 19 and that phosphomimetic mutations at this position produce a gpsB null-like phenotype both in B. subtilis and in L. monocytogenes. Similarly, phosphoablative mutations have no demonstrable effect in either species. The straddling of T88 by F91 and L94 suggests that the interaction between GpsB and PrkC will overlap with the interface between C-GpsB subunits, and hence, the interaction with PrkC could adjust the arrangement of subunits in the hexamer. The interaction with PrkC could therefore, in turn, influence the interaction with PBP1 by altering the GpsB quaternary structure and the arrangement of PBP1 binding sites. However, the phosphorylation of GpsB, mediated by PrkC, is not in itself sufficient to affect the interaction with PBP1 as both wild-type and phosphomimetic variants of GpsB behaved identically when combined with BsPBP1 in activity assays (Fig. 8). We have shown here and elsewhere 11 that correct hexamer formation is essential for GpsB function in L. monocytogenes, which invites the suggestion that the phosphomimetic mutation instead affects GpsB function by altering its quaternary structure. However, a major effect on GpsB quaternary structure was not evident here as the two phosphomimetic mutants of BsGpsB, T75D and T75E, behaved indistinguishably from wild-type BsGpsB on SEC and had an identical thermal stability on circular dichroism analysis. Perhaps the gpsB null phenotype of phosphomimetic mutations reflects the introduction of a net negative charge in this region affecting the ability of GpsB to interact with PrkC or another divisome component. A molecular rationale for the gpsB null phenotype associated with the phosphomimetic mutation reported earlier 19 and herein remains to be determined.
An important functional consequence of a closed tripod-like arrangement, as opposed to a more open “splayed out” arrangement of subunits, is the potential for the GpsB hexamer to interact simultaneously with multiple membrane-embedded PBP1 molecules. In the SAXS model of GpsB, the binding sites for PBP1 (a surface cleft in the N-terminal domain 11 ) are clustered at one end of the elongated hexamer (Fig. 9). Similarly, the residues critical for the association of GpsB with the cell membrane (L24 and R25 in LmGpsB 11 ) are also clustered at the same end of the hexamer (Fig. 9). GpsB, therefore, has the implicit capacity to interact with multiple PBP1 molecules and thus to impact upon the arrangement of PBP1 molecules in the divisome, and this capacity may explain phenotypic and genetic evidence that GpsB regulates cell wall synthesis.5,11 In E. coli, the essential cell division protein FtsN, which is unrelated to GpsB, may play a similar role as it stabilizes dimeric or multimeric forms of PBP1B in vitro. 48
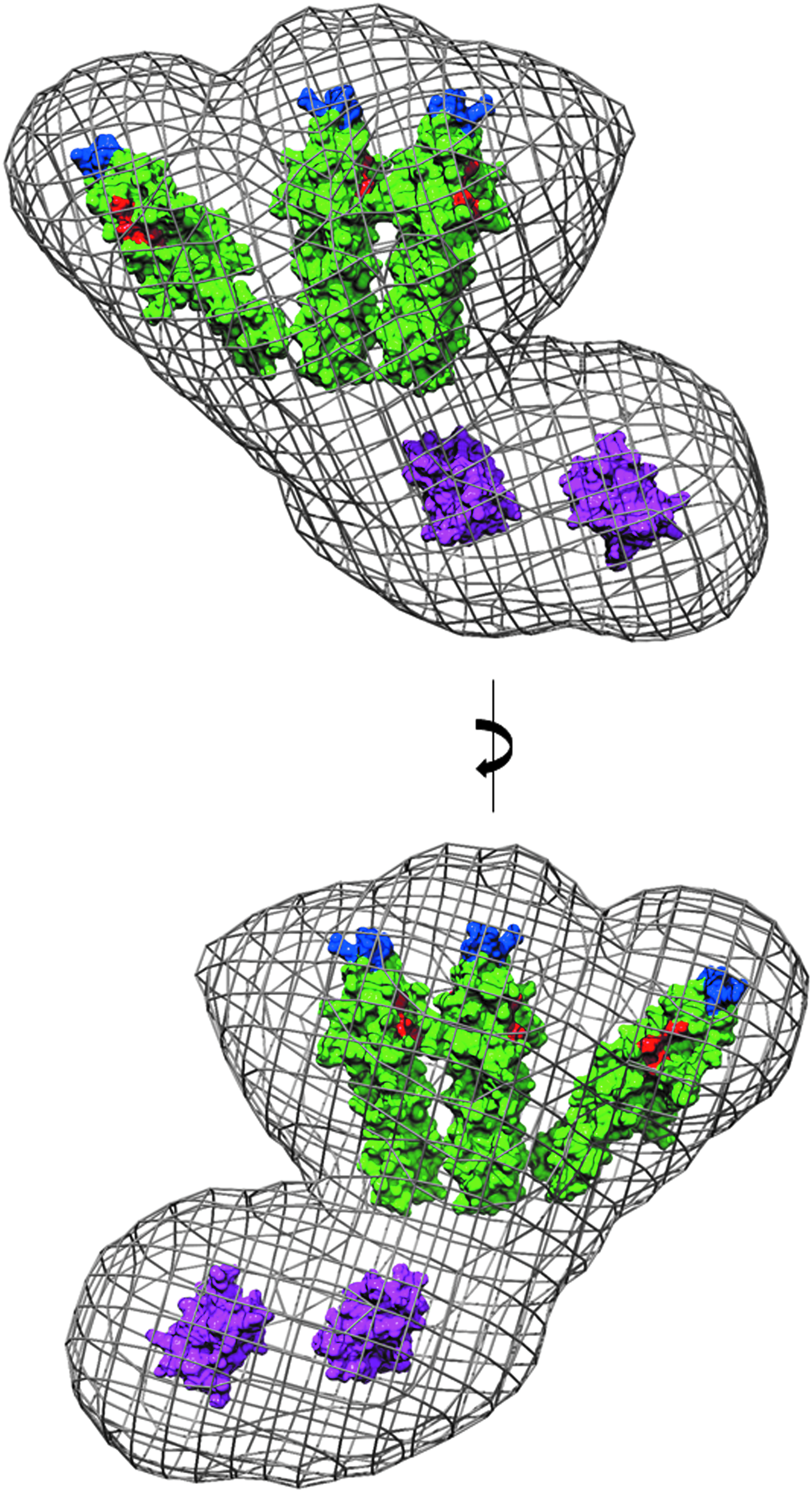
PBP1 and membrane binding sites in the SAXS model of GpsB. The atomic coordinates of N-GpsB dimers and C-GpsB trimers, represented as molecular surfaces (green and magenta, respectively), have been manually positioned in the averaged ab initio molecular envelope of the LmGpsB hexamer, shown as a gray mesh. Surface exposed residues critical for the interaction of LmGpsB with PBP111 are colored red (D33, D37, I40). Residues critical for association of LmGpsB with the cell membrane 11 are colored blue (L24, R25). The molecular envelope represented here is the average of 15 independently generated ab initio dummy atom models.
At 37°C, the ΔgpsB null mutant makes the overexpression of PBP1 lethal in L. monocytogenes, 11 which suggests that GpsB acts as a negative regulator of at least one of the enzymatic activities of PBP1. A simple regulatory mechanism could entail GpsB promoting the oligomerization of PBP1, a feasible scenario given that the GpsB hexamer has the potential to interact with multiple PBP1 molecules. PBP1B from E. coli forms dimers, and dimerization enhances both GTase and TPase activities in vitro. 41 The activities of E. coli PBP1B are stimulated or modulated by interactions with LpoB, FtsN, TolA, and CpoB, and these interactions are essential for the function of this peptidoglycan synthase in the cell.39,48–53 However, there was no effect of BsGpsB on either the GTase or the TPase activity of PBP1 in vitro (Fig. 8), which would argue against a regulatory mechanism. Any modulation of PBP1 activity by GpsB in vivo must therefore require other divisome components and/or membrane-bound elements that are not recapitulated in these experiments in vitro. In particular, simply by controlling the spatial arrangement of PBP1 molecules in the membrane in vivo, GpsB may influence both the arrangement of peptidoglycan strands and the pattern of intrastrand cross-links in the cell wall.
In this regard, the hexameric GpsB, and its flexible nature, could potentiate PBP1 activity or cellular localization by changes to the quaternary structure of GpsB: movements of the GpsB domains relative to one another could translate to large changes in the spatial arrangement of PBP1 binding sites. GpsB might thus act as an allosteric sensor, with its interaction with PBP1 modulated by changes in quaternary structure induced by interactions with other purported binding partners such as PrkC, EzrA, MreC, or DivIVA. These ideas will form the focus of our future experiments.
Footnotes
Acknowledgments
The authors thank Dr. Stephen Prince at the University of Manchester for advice on planning SAXS experiments. They thank Dr. Owen Davies at the University of Newcastle for SEC-MALS analysis. They would also like to thank PETRAIII and Diamond Light Source for beamtime on beamlines P12 and B21, respectively, and Drs. Cy Jefferies and Mark Tully for assistance during SAXS data collection. They thank Peter Sharratt at the Peptide and Nucleic Acid Facility, Cambridge University, for amino acid analysis. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under BioStruct-X (grant agreement #283570 to CB for SAXS beamtime), the UK Biotechnology and Biological Sciences Research Council (BB/M001180/1 to RJL), the Wellcome Trust (101824/Z/13/Z to WV), and the German Research Foundation (HA 6830/1-1 to SH).
Disclosure Statement
No competing financial interests exist.