
Abstract
Objective:
The aim of this work was to purify and characterize a bacteriocin-like antimicrobial substance produced by an antagonistic active strain of Enterococcus faecium.
Methods and Results:
A novel bacteriocin-like inhibitory substance (BLIS) produced by the E. faecium ICIS 8 strain was purified and characterized using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and N-terminal amino acid sequencing revealed the following partial sequence: NH2-APKEKCFPKYCV. The proteinaceous nature of purified BLIS was assessed by treatment with proteolytic enzyme. Studies of the action of BLIS using bacteriological and bioluminescence assays revealed a dose-dependent inhibition of Listeria monocytogenes 88BK and Escherichia coli K12 TG1 lac::lux viability. The interaction of the BLIS with the bacterial surface led to the compensation of a negative charge value, as shown by zeta-potential measurements. Assessments of membrane integrity using fluorescent probes and atomic force microscopy revealed the permeabilization of the cellular barrier structures in both L. monocytogenes and E. coli.
Conclusion:
The novel BLIS from E. faecium ICIS 8 was characterized by a unique primary peptide sequence and exerted bactericidal activity against L. monocytogenes and E. coli by disrupting membrane integrity.
Introduction
B
Bacteriocins are ribosomally synthesized bacterial peptides with antimicrobial activity that provide intermicrobial antagonism and are involved in cell-to-cell communication. 3 The bacteriocins produced by enterococci are called enterocins. Bacteriocin-producing strains have been isolated from a variety of sources, from animal intestinal microbiota to mangrove forests.4,5 The primary structure of enterocins places most of these compounds in bacteriocin class II.4,6 These bacteriocins are characterized by a relatively narrow spectrum of antimicrobial activity that is predominantly related to Gram-positive bacteria from closely related species.6–8
The primary mechanism of action of bacteriocins includes pore formation and/or the inhibition of cell wall biosynthesis. 7 The initial interaction of bacteriocin with bacterial cells occurs through electrostatic forces. The subsequent processes are mediated by the interaction with receptors, such as lipid II, 3 the permease mannose-phosphotransferase, 7 which increases an affinity of bacteriocin molecule by several times. 9 The followed events include hydrophobic interaction of peptide with phospholipids of membrane leading to permeabilization of bacterial cell. Bacteriocins also can realize their action by the target-independent pathway, 10 which is similar to the mechanism of action of magainin, a cationic peptide produced by eukaryotic organisms. 11 However, it should be noted that the specific mechanism of action depends on many factors, the most important of which is the active concentration of the bacteriocin. 12 In addition, it is currently unclear whether bacteriocins require receptor binding for their activity. 13
Although the effects of bacteriocins produced by a lactic acid bacteria (LAB) on Gram-positive bacteria have been studied, questions related to the action of these peptides against Gram-negative bacteria have been poorly investigated. 14 Available data suggest the possibility of bactericidal action of LAB-bacteriocins against various Gram-negative species.15–17 In some cases, activity of enterocins showed only upon the additional treatment of Gram-negative bacteria by osmotic shock, temperature changes, or chelating agents.18,19 Thus, the LAB-bacteriocin activity against phylogenetically distant species and the mechanism of action at the molecular and cellular levels against Gram-negative bacterial strains remains to be a little studied.
The detailed analysis of action of bacteriocins produced by a Gram-negative bacteria showed a diverse mechanism of action against same bacteria, for example, colicin E2 has enzymatic activity and target nucleic acid, 20 while colicin E1 has a pore forming activity. 13
In this regard, the aim of this work was to purify and characterize an antimicrobial substance that could be a bacteriocin-like inhibitory substance (BLIS) produced by an antagonistic active strain of Enterococcus faecium ICIS 8. This Enterococcus strain was isolated from the human intestine and is characterized by the presence of the antagonistic activity against Listeria and the absence of phenotypic manifestation of the pathogenicity factors (DNase, gelatinase, and hemolytic activity). 21 Also in this work, the features of the biological action of the purified BLIS against Listeria monocytogenes 88BK and Escherichia coli K12 TG1 were studied.
Materials and Methods
Isolation and characterization of the BLIS
The E. faecium ICIS 8 was cultured in 300 ml of Schaedler broth (HiMedia) for 24 hrs at 37°C. Cells were removed from the growth medium by centrifugation (20 min, 4°C, 9,000 g). Protein concentration was estimated by the Lowry method as described in. 22
At the next step, the desalting of culture medium was performed using a reversed-phase low pressure chromatography on Brownlee Aquapore RP-300 column (PerkinElmer) equilibrated with solvent A (10% acetonitrile in ultra purified water (Milli Q) with 0.1% trifluoroacetic acid [TFA]). Elution was performed using the solvent B (80% acetonitrile in ultra purified water with 0.1% TFA). Then the concentrate was evaporated to remove acetonitrile by a Labconco concentrator followed by lyophilization to withdraw a residual quantity of TFA. The obtained desalted extract was tested to reveal the antibacterial properties. The fractions were separated by size exclusion high pressure liquid chromatography (SE-HPLC) method on a BioSep-SEC-s2000 (7.8 × 300 mm) column (Phenomenex) integrated with HPLC system “Knauer Smartline” (Germany). Elution was carried out by 12% acetonitrile in ultra purified water with 0.1% TFA, at a flow rate of 0.6 ml/min. The peptide fractions were monitored by measurement of absorption at 214 nm and collected manually. After that, the fractions collected were completely evaporated, as described above, and dissolved in distilled water to check for presence of antimicrobial properties by well agar diffusion assay. The second step of purification was performed by reversed-phase high pressure liquid chromatography (RP-HPLC) on a Luna C18 (4.6 × 150 mm) column (Phenomenex). Peptides were eluted from the column with a linear gradient of acetonitrile in aqueous 0.1% (v/v) TFA as: 10–50% for 25 min, 50–85% for 25–50 min, and 85–100% for 50–55 min at a flow rate of 0.9 ml/min. The peptides were detected at 214 nm. Fractions were collected manually, also fully evaporated, as described above, and checked for antimicrobial activity.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis of active fraction
The electrophoretic separation of the active fraction obtained by RP-HPLC was conducted according to the protocol described. 23 To an analyzed sample, an equal volume of buffer containing 0.125 M Tris-HCl, pH 6.8, 4% sodium dodecyl sulfate (SDS), 20% glycerol, and 10% 2-mercaptoethanol was added. One milliliter of a 0.2% solution of bromophenol blue was mixed with the sample and applied to the wells of the polyacrylamide plate consisting of 4% in the stacking gel and 10% in the separating gel. The phosphorylase B (97 kDa), bovine serum albumin (66 kDa), ovalbumin (45 kDa), carbonic anhydrase (30 kDa), trypsin inhibitor (20.1 kDa), and α-lactalbumin (14.4 kDa) (Amersham Biosciences) were used as the markers of molecular mass of the proteins.
After the electrophoresis (1.5–2.0 hr), gel was transferred to PVDF membrane “Immobilone.” The membrane was stained with 0.1% solution of Coomassie R-250 in 50% aqueous methanol for 3–5 min and washed with water.
N-terminal sequencing
The peptide was sequenced by automated Edman degradation on a model PPSQ-33A Shimadzu sequencer (Shimadzu Corp.) according to the manufacturer's protocol. Cysteine residues were determined as pyridilethylated derivatives. Homology search was performed using UniProt/Swiss-Prot databases with a BLASTP algorithm.
Effect of enzyme and temperature treatment on BLIS activity
The protease susceptibility was carried out by incubation of BLIS with 1 mg/ml of protease K (Sigma-Aldrich) at 37°C for 1 hr in buffer (10 mM Tris-HCl, pH 7.5, and 1 mM CaCl2·H2O) as recommended by the suppliers. BLIS in buffer without enzyme was used as positive control; enzyme in buffer solution and buffer without any additives were used as negative controls. Evaluation of thermostability of BLIS was performed by heating to 60°C/30 min and 80°C/30 min in solid state thermostat “Thermit” designed for microfuge tubes (“DNK-Technologiya”). The inhibitory activity of BLIS after treatments was determined using the well agar diffusion assay.
Determination of activity by well agar diffusion assay
Determination of bactericidal properties of fractions collected was performed by a well agar diffusion assay. 24 The microorganism indicator L. monocytogenes 88BK was cultured for 18 hr in Schaedler broth (HiMedia), after which 100 μl of the bacterial suspension (containing ∼107 CFU) was mixed with 10 ml of soft (0.5%) Schaedler agar, and placed immediately over the Petri dish, which was previously overlaid with 1.5% Schaedler agar plate. Solidified agar plates were punched with a 5-mm diameter flame-sterilized cork borer, and the serial twofold diluted fractions (10 μl) were transferred into the wells. After incubation at 37°C overnight, inhibitory areas were observed.
Evaluating of antimicrobial activity of the purified BLIS
For evaluating the BLIS action, a Gram-positive strain of L. monocytogenes 88BK and Gram-negative strain of E. coli K12 TG1 were taken. Bacteria were cultured on Schaedler broth (HiMedia) overnight at 37°C, where a final bacterial concentration of 3 × 108 CFU/ml was reached (OD670 0.1). Bacterial suspensions were diluted to achieve a cell amount of 3 × 105 CFU/ml for bacterial growth monitoring. Affecting of bacterial growth by BLIS was evaluated by a microdilution broth assay in 96-well microtiter plate against L. monocytogenes 88BK and E. coli K12 TG1 as described in protocol. 25 The bacterial suspension was incubated with twofold dilutions of the BLIS, and dynamic of bacterial growth was assessed by reading and plotting the absorbance data at 620 nm obtained by spectrophotometer IEMS MF (Labsystems). Antimicrobial activity of BLIS was expressed by the minimal inhibitory concentration (MIC), which was defined as the lowest peptide dose at which no visible growth was detected.
For evaluating the medium composition on BLIS action, the separate experimental groups were formed. Each group consisted of L. monocytogenes or E. coli cells (3 × 105 CFU/ml) were mixed with 3.7 μg/ml of the BLIS in the ultra purified water. The BLIS action was evaluated separately at the presence of 1, 5, and 10 mg/ml of bovine serum albumin (Sigma-Aldrich) or NaCl at 0, 75, and 150 mmol. After incubation for 1 hr, the bacterial suspensions were plated on Schaedler agar for cultivation and followed CFU counting.
The bioluminescence assay
The bioluminescence inhibition assay was performed as described (Efremova et al. 2015). Briefly, lyophilized E. coli K12 TG1 lac::lux cells purchased from “Immunotech” were rehydrated with cold distilled water to a concentration of 3 × 107 CFU/ml, exposed for 30 min at 2–4°C, and then the temperature was raised up to 25°C before application. For bioluminescence assay, the 20 μl of bacterial suspension was mixed with appropriate volume of twofold dilutions of the BLIS and placed into a well-plate of a “Microlite 2+” microplate with nontransparent side walls (Thermo) up with sterile deionized water to a final volume of 100 μl. The wells were filled with sterile deionized water and contained an appropriate amount of bacterial biosensor used as the negative control; the wells containing bacterial biosensor mixed with various concentrations of nisin (Sigma-Aldrich) were used as the positive controls. Bioluminescence measurements were carried out using an LM-01T microplate luminometer (Immunotech), which dynamically registered the luminescence intensity of the samples for 60 min, estimated in relative light units (RLU). The data were analyzed using KILIA graphing software provided with the instrument. To quantify the bioluminescence inhibition index (I) due to toxicity of the BLIS, we used the algorithm
Zeta-potential measurements
The test microorganisms were grown in Schaedler broth (37°C, 12 hr). Bacterial cells were precipitated by centrifugation (5,000 g for 10 min) and resuspended in ultra purified water to achieve a cell amount of 1 × 106 CFU/ml followed by incubation with the BLIS at several concentrations for 10 min at 37°C. The zeta-potential measurements of bacteria were performed with Fotocor Compact Z (Fotocor) and calculated according to bacterial cell electrophoretic mobility using the Smoluchowski equation.
Fluorescent spectroscopy
For fluorescent staining, LIVE/DEAD BacLight Bacterial Viability Kit (Molecular Probes) was used according to manufacturer's protocol. Bacterial cells were precipitated by centrifugation at 5,000 g for 5 min; the pellet obtained was resuspended in 1 ml ultra purified water to receive OD670 ∼0.03 and incubated with several concentrations of the BLIS for 1 hr. Fluorescence detection was performed by fluorescent spectrometer Solar SM 2203 (Republic of Belarus). The ultra purified water and 70% isopropanol were used as a negative and positive control, respectively. The percentage of bacteria with intact/damaged membranes was calculated according to the green/red fluorescence signal ratio.
Atomic force microscopy
Using an atomic force microscopy (AFM), a morphofunctional reaction of Gram-positive and Gram-negative bacteria on the BLIS action was investigated. The test microorganisms were grown in Schaedler broth (37°C, 12 hrs). Bacterial cells were precipitated by centrifugation (5,000 g for 10 min) and resuspended in ultra purified water to achieve a cell amount of 1 × 106 CFU/ml followed by incubation with the BLIS at several concentrations for 30 min at 37°C. After incubation with the BLIS, the cells were washed by centrifugation (5,000 g for 10 min) and resuspended in ultra purified water. The observations were obtained with an atomic force microscope SMM-2000 (JSC Proton-MIET Plant) in contact mode in air. The instrument was equipped with silicon nitride cantilever MSCT-AUNM (Veeco Instruments, Inc.) with a pyramidal (V-shaped) tip with a typical radius of ∼10 nm. During AFM imaging, the dimensional features and the root-mean-square roughness (Rrms—the standard deviation of the Z values) for the height images were conducted over 5 × 5-μm areas on different surfaces and were calculated using SMM-2000 software. The volume of the cells was calculated according to. 26
Statistical analysis
Experiments were performed using three independent series. The values were expressed as mean ± standard deviation. Statistical manipulation of obtained results was realized with STATISTICA 6.0 (StatSoft, Inc.) software. To estimate difference significances, the Wilkinson paired test was carried out. Differences were significant at p < 0.05.
Results
Purification and characterization of the BLIS
The first stage of the purification of desalted growth medium is to separate the compounds that are localized in the culture medium. To achieve this goal, we used SE-HPLC. The results are presented in Fig. 1a. All collected fractions were tested for antimicrobial activity, and only the fraction that was eluted first (14.98 min after injection) displayed an antimicrobial effect. Based on the retention times of the main protein standards (ovalbumin, 45 kDa; RNase A, 13.7 kDa; and insulin, 5.8 kDa) (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/mdr) during the SE-HPLC, we propose that the molecular mass of the active polypeptide is ∼10–20 kDa. The second purification step included the separation of the active fraction by RP-HPLC, which yielded five fractions (Fig. 1b). Among the collected and tested fractions, only one fraction could inhibit Listeria. Subsequently performed sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of the active fraction showed that the main protein band belongs to a mass interval from 10 to 14 kDa (Fig. 1c). This fraction appeared to be heat stable and completely inactivated by protease treatment (Table 1).
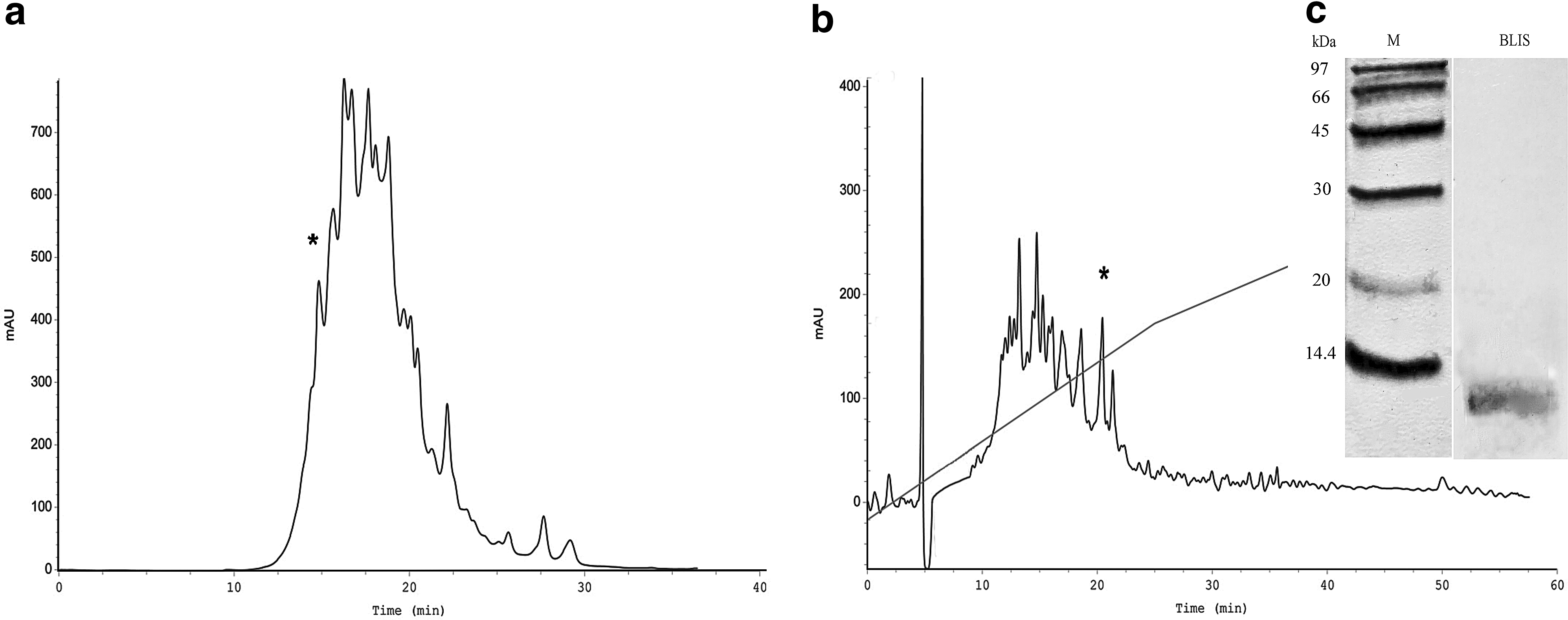
Purification of BLIS by two-step separation based on HPLC procedures:
N-terminal amino acid analysis of the purified antimicrobial polypeptide revealed the following partial sequence: NH2-APKEKCFPKYCV. A subsequent search for protein homology using the NCBI databases through the BLASTP algorithm did not reveal any similarity to other known bacteriocins.
Growth dynamics of Gram-positive and Gram-negative bacteria during exposure to the BLIS
The growth of the indicator strain L. monocytogenes 88BK was completely inhibited by the BLIS at 3.7 μg/ml, which was determined to be the MIC (Fig. 2a). In contrast, the addition of the BLIS to the growth medium with E. coli K12 TG 1 led to the complete suppression of growth at 30 μg/ml, which was determined to be the MIC (Fig. 2b).
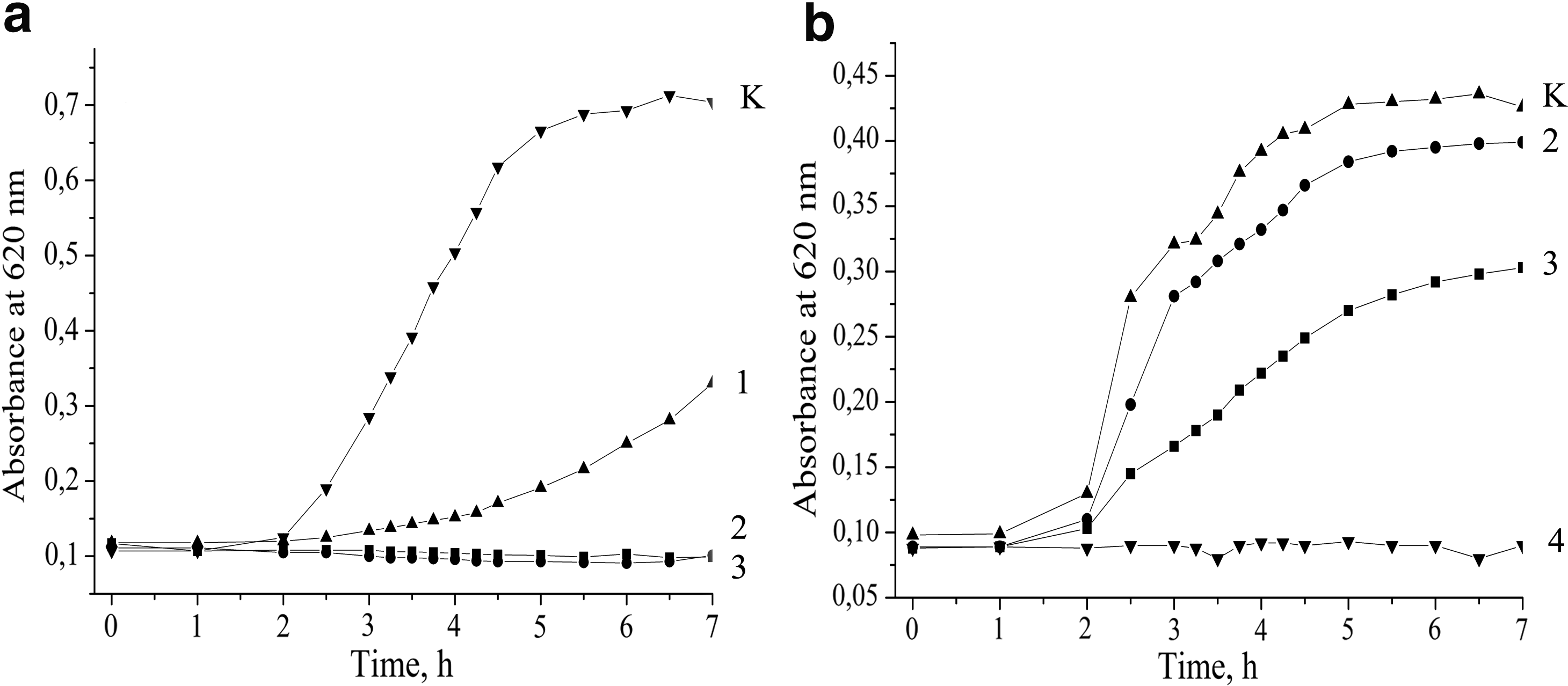
The growth kinetic of Listeria monocytogenes 88BK
Influence of the BLIS on bacterial bioluminescence
A dynamic of the manifestation of the BLIS toxic effect was assessed using the E. coli K12 TG1 lac::lux biosensor. Coincubation of the BLIS with E. coli cells led to a dose-dependent effect on bacterial bioluminescence. Specifically, the incubation of E. coli with the highest concentration of the BLIS resulted in the complete inhibition of bioluminescence at the 10 min time point (Fig. 3a). E. coli cells that were treated at lower concentrations showed significantly reduced, but stable bioluminescence kinetics. Based on the obtained bioluminescence data, the EC50 toxicological parameter was calculated to be 1.7 μg/ml.
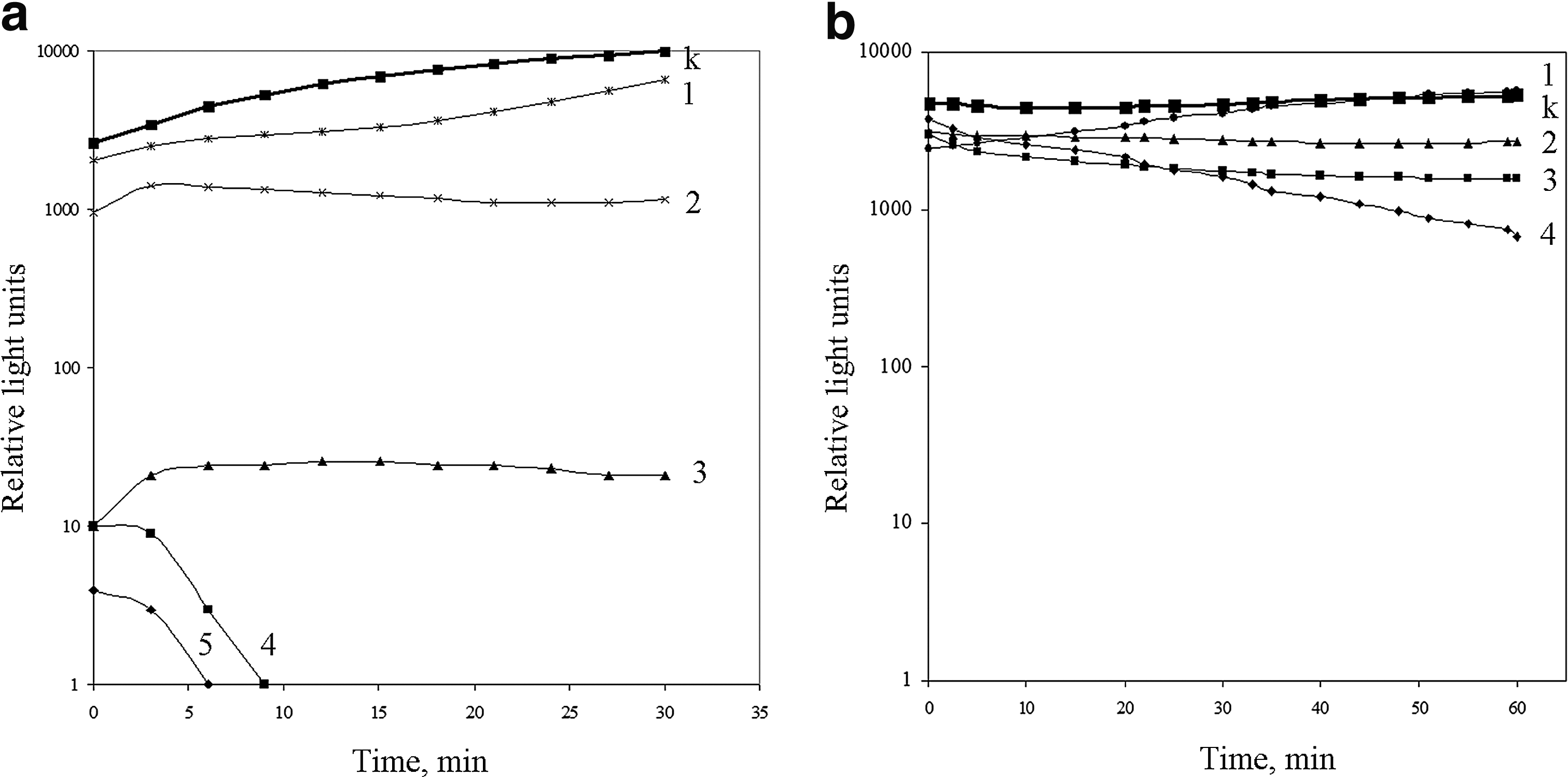
The kinetics of E. coli K12 TG1 lac::luxCDABE luminescence under the BLIS
The treatment of E. coli biosensor by nisin also revealed dose-dependent progress-in-time inhibition of bioluminescence (Fig. 3b). The EC50 toxicological parameter was calculated to be 2.7 μg/ml.
Influence of the ionic strength and protein content of the medium on the antimicrobial activity of the BLIS
The results obtained from the growth kinetics and bioluminescence inhibition assays led us to determine the factors that modulate the antimicrobial activity of the BLIS in various environments. First, the antimicrobial activity of the BLIS was evaluated in the presence of 0–150 mmol of NaCl. The viability of both strains was significantly inhibited by the BLIS at various salt concentrations (Fig. 4a, c). The second assessed factor was bovine serum albumin, which was investigated at concentrations of 1–10 mg/ml. The presence of this protein did not significantly affect the activity of the BLIS against Listeria (Fig. 4b), but the observed effect on E. coli cells was completely inhibited by 10 mg/ml of BSA (Fig. 4d).
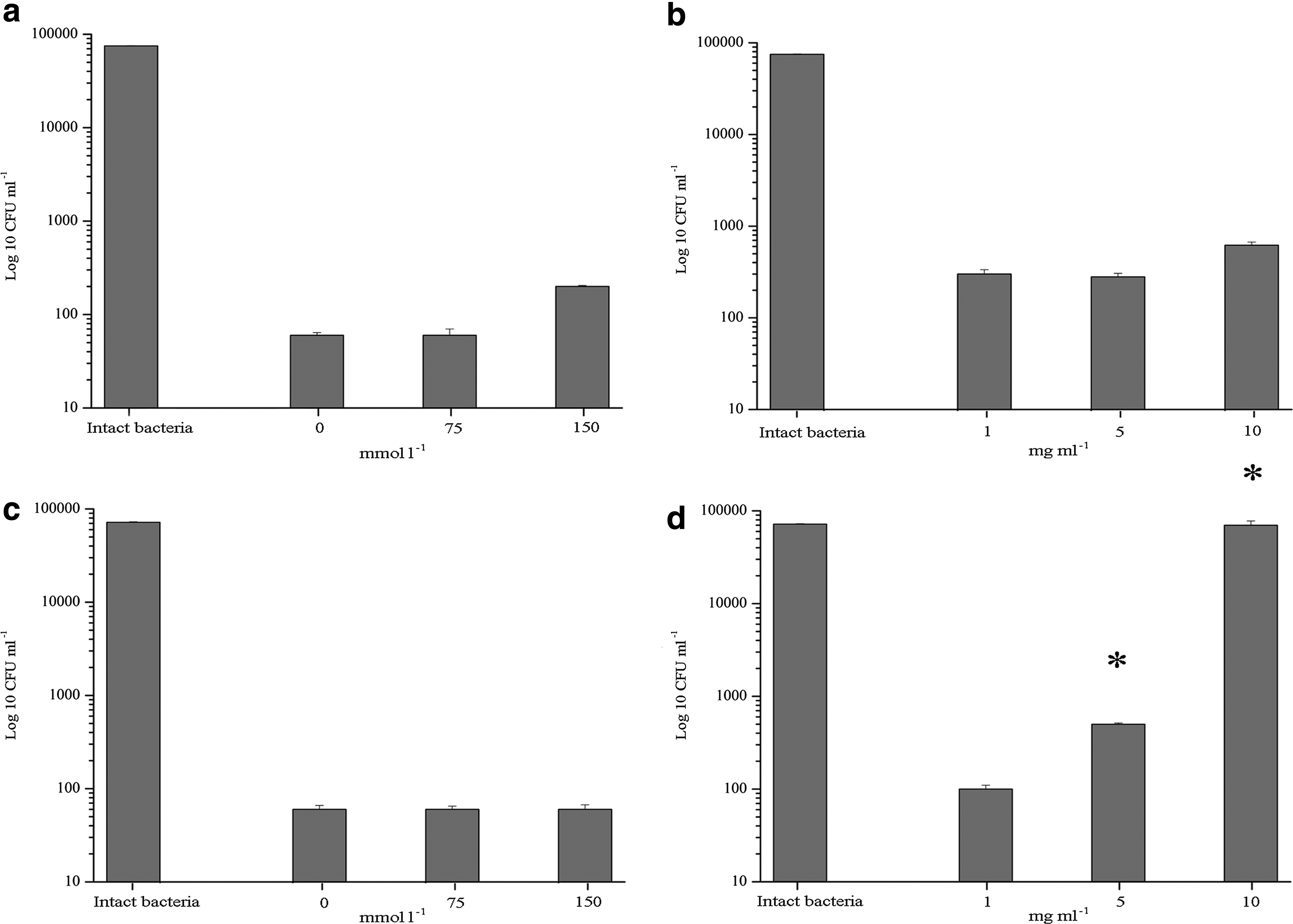
Effect of ionic strength
The interaction of the BLIS with bacteria evaluated using zeta-potential and membrane integrity measurements
The addition of the BLIS to the suspension of L. monocytogenes at a concentration of 3.7 μg/ml shifted the zeta-potential from −31 ± 1.0 to −14 ± 1.5 mV, but the addition of further BLIS did not change the zeta-potential (Fig. 5a). The intact E. coli cells were characterized by a zeta-potential value of −38 ± 0.3 mV, and the addition of the antimicrobial substance led to negative surface charge neutralization in a dose-dependent manner until a final value of −16 ± 1.0 mV was reached (Fig. 5b).
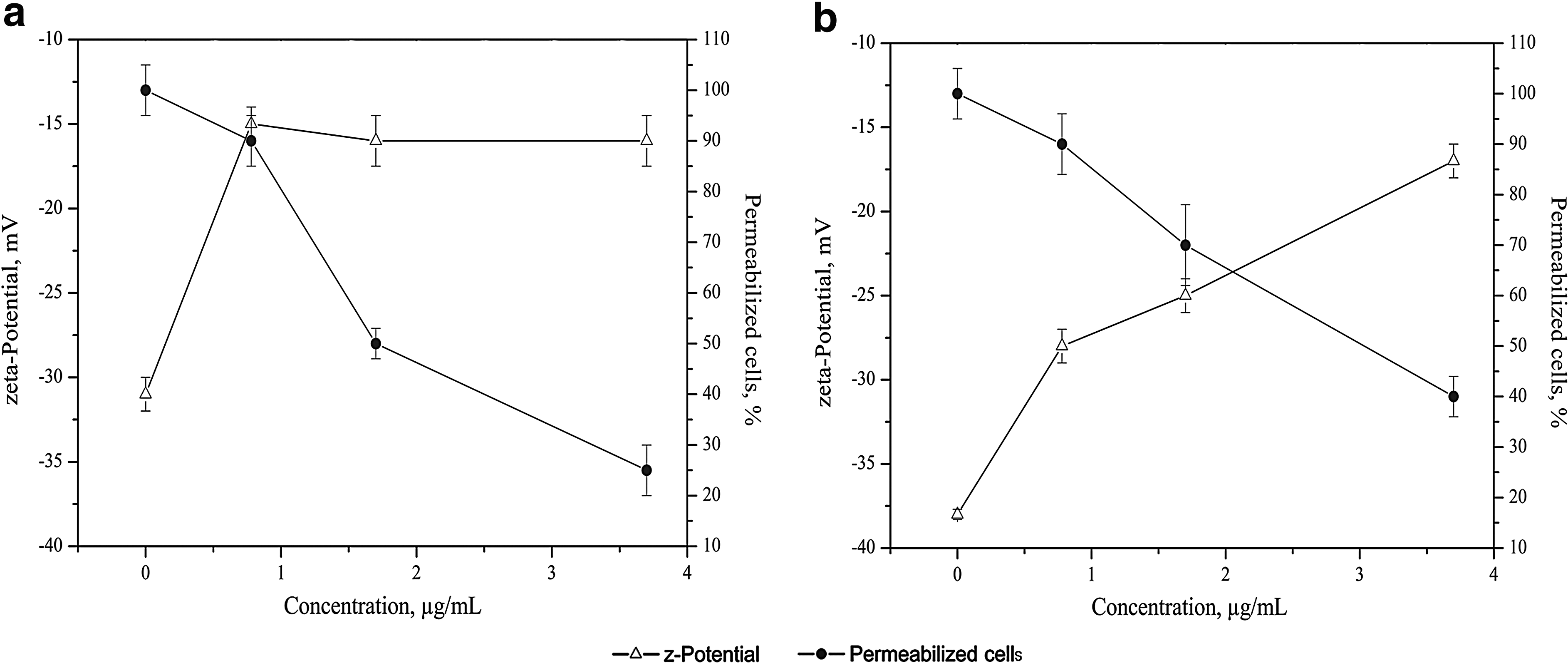
Effect of the BLIS on bacterial zeta-potential and cell membrane integrity. Interaction of the peptides with L. monocytogenes 88BK
The permeability of the cells, which was evaluated using LIVE/DEAD staining, was significantly increased during exposure to the BLIS. Specifically, 50% of the Listeria cells in the treated population were affected when the BLIS was added at the MIC (Fig. 5a). The fluorescence permeabilization assay revealed the relative resistance of E. coli cells to the effects of the BLIS. Disrupting the permeability of 50% of cells in the E. coli population required a twofold excess of the BLIS in comparison to Listeria cells (Fig. 5b).
The morphofunctional reaction of bacterial cells treated with the BLIS
AFM of intact Listeria cells allowed us to characterize their volume as 0.70 ± 0.17 nm3 and revealed that the topography of the bacterial surface was relatively smooth (Fig. 6a), with a calculated mean-square roughness of the surface (Rrms) of 1.24 ± 0.33 nm. In contrast, the calculated cellular volume of intact E. coli (Fig. 6b) was 1.99 ± 0.49 nm3, and the cell surface had a roughness equal to 2.35 ± 0.45 nm.
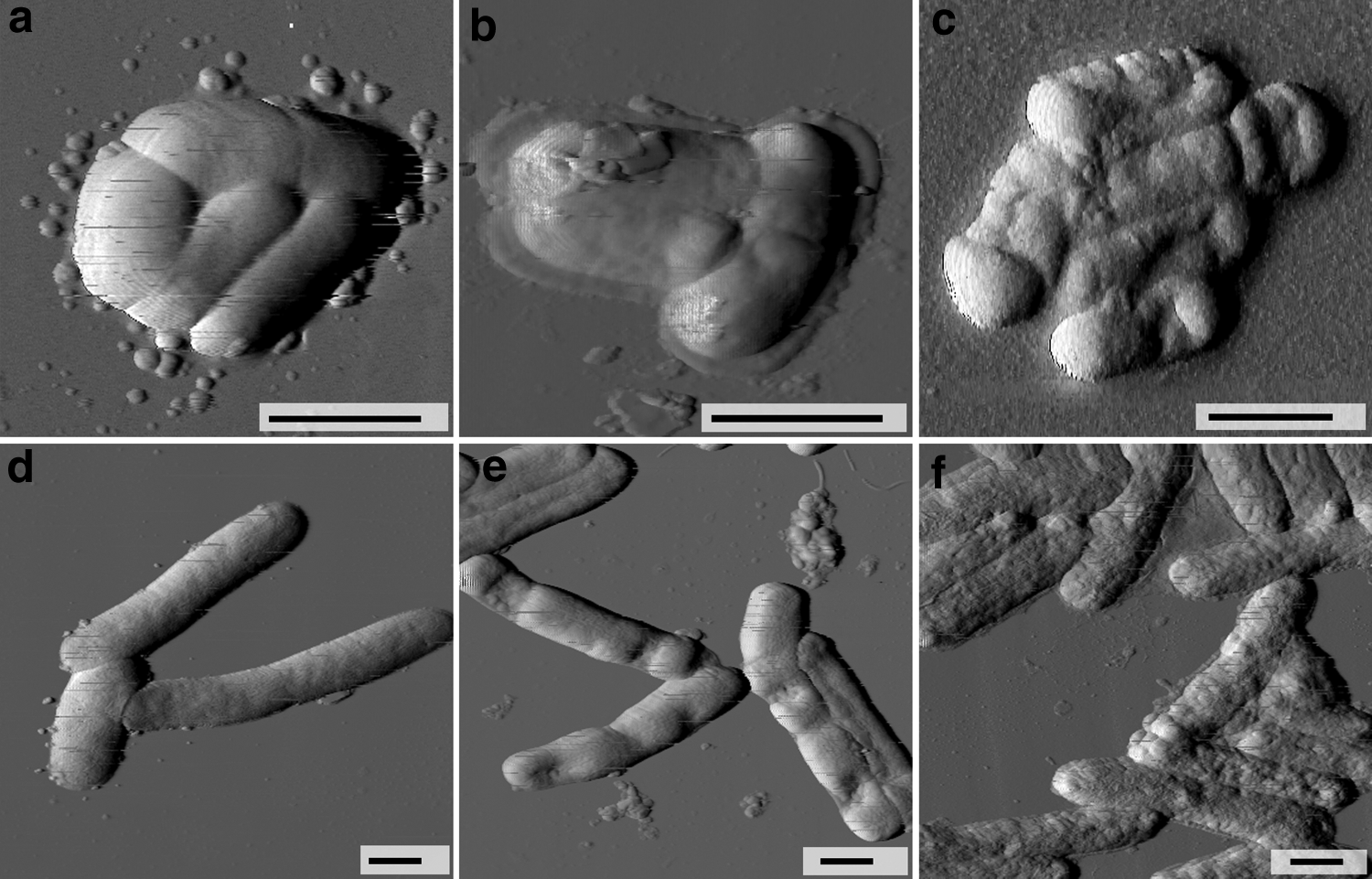
Effect of the BLIS action on bacterial morphology. AFM images of the intact bacterial cells of the L. monocytogenes 88BK
The treatment of Listeria with the BLIS at the MIC did not lead to a significant disturbance of the cells (Fig. 6b). The bacteria retained their volume and surface properties, with measured volume and roughness values of 0.67 ± 0.29 nm3 and 1.37 ± 0.32 nm, respectively. These values did not differ from the parameters of the intact bacteria (p > 0.05). Significant changes were observed (Fig. 6c) after treatment with a BLIS concentration twofold higher than the MIC. Most of the observed cells exhibited a loss of cell volume, with pronounced signs of cell wall destruction. Quantitatively, the bacterial changes were expressed as a reduction of the cellular volume to 0.365 ± 0.14 nm3 (p < 0.05) and an increase in surface roughness to a value of 12.6 ± 4.5 nm (p < 0.05). The morphofunctional reaction of Gram-negative E. coli to the BLIS was similar to that observed for Listeria. In particular, AFM images of E. coli treated with 3.7 μg/ml of the BLIS revealed localized collapsed areas of the cellular surfaces (Fig. 6e) and the reduction of the volume to 1.34 ± 0.37 nm3 (p < 0.05). However, the surface morphology was not affected (except for the collapsed areas), as the measured roughness was 2.27 ± 0.59 nm (p > 0.05). Additional results were obtained when the BLIS concentration was increased to 7.4 μg/ml, which led to the reduction of the cellular volume to 1.32 ± 0.42 nm3 (p < 0.05) and significant changes in the surface ultrastructure. AFM detected a loosened outer membrane (Fig. 6f), with a roughness value that was increased to 6.22 ± 1.00 nm (p < 0.05).
Discussion
In a previous study, the morphological reaction of L. monocytogenes 88BK cells treated with culture medium from E. faecium ICIS 8 was investigated using AFM. 27 Continued work in this direction involved studies of the biological activity of the antimicrobial polypeptides that were purified from the culture medium of E. faecium ICIS 8.
Using a two-step purification process that included size exclusion and reversed-phase high pressure chromatography, the polypeptide fraction with antimicrobial properties was isolated. This fraction contained the polypeptide that was eluted in the first peak groups by SE-HPLC separation. This finding suggests that the active factor is a protein rather than a peptide. The proteinaceous nature of active factor confirmed by the treatment with the proteinase K revealed the sensitivity to proteolytic enzyme. Further purification by RP-HPLC using the C18 hydrophobic phase allowed us to collect a peak that was subsequently analyzed using electrophoresis. Performed SDS-PAGE of the active fraction showed that the protein band belongs to a mass interval from 10 to 14 kDa. The N-terminal amino acid sequence of the native rechromatographed compound allowed us to determine only 12 amino acid residues, including two cysteine and two proline residues. The data suggested that the BLIS has a partial amino acid sequence that differs from those of other known bacteriocins, particularly those from the Enterococcus genus.
The biological action of the BLIS against bacteria proceeds according to well-described phases of the interaction of these molecules with bacterial cells. At the first stage, the electrostatic sorption of peptides on bacterial surface occurred. It was confirmed by measurements of the zeta-potential, which revealed a fast-moving compensation of the negative cell wall charge during exposure to the BLIS. The followed events include disturbance of the membrane integrity of bacteria. It was detected through fluorescence spectroscopy using the DNA-binding dyes SYTO 9 and propidium iodide for both Listeria and E. coli cells. The differences observed between the species were explained by the concentration of BLIS that led to cell permeabilization; in particular, this concentration was higher for E. coli. The structural changes of bacterial cells also were studied using high-resolution microscopy techniques, such as AFM. This technique can recognize and quantitatively describe the peptidoglycan disruption of Gram-positive bacteria 28 and the destabilization of the outer membrane of Gram-negative bacteria29,30 in response to application of antimicrobial peptides. Using AFM, changes in the normal shape of Listeria cells under the influence of the BLIS were detected, and a decrease in bacterial volume occurred after treatment. The outer membrane of E. coli cell wall was also a target for the action of BLIS, as revealed by AFM. The typical signs of antimicrobial peptide action,20,31 such as pore-like lesions and flattening at the apical end of the cells, were detected. All these features of the BLIS action are consistent with the data on the activity of other enterocins. This thesis well illustrates the work 32 where the cationic enterocin ST4SA at low concentration caused the formation of pores and leakage of small intracellular molecules and efflux of intracellular macromolecules at higher dose. Similar results were reported for enterocin CRL35 33 and enterocin E-760. 15 Recently was reported about a novel pore-forming enterocin LD 3 with activity against E. coli. 34
To investigate how bacteria respond to treatment at population level, the classic method of culturing bacteria in the presence of various concentrations of BLIS was used. Measurements of the optical density of the cell population during incubation with the BLIS showed a slight inhibition of E. coli growth at some time points, while the growth of Listeria was affected significantly more strongly. This result is similar with data concerning action of the enterocin AS-48 that can reduce the number of viable E. coli K12 cells at significantly (150 μg/mL) much higher concentration than those used for inhibition of Gram-positive bacteria Listeria sp. (0.1–2.3 μg/mL). 34
In light of these results, we decided to study this polypeptide action in a more detailed manner, specifically against relatively resistant E. coli. For this reason, we used a more sensitive method of bacterial biosensor for toxicological studies. The luminescence inhibition assay allows us to recognize nanomolar concentrations of toxic compounds, and the process is more rapid than traditional bacteriological methods, such as agar plating and growth monitoring.35,36 Using the bioluminescence assay, the rapid inhibition of bacterial luminescence was observed within the first minutes of the assay in a dose-dependent manner. Because the bacterial luciferase complexes associate with the bacterial membrane, 37 the effects on these structures are the starting point of subsequent luminescence inhibition. Moreover, the luminescence of E. coli containing the luxCDABE cassette depends on energy metabolism within the cell, and the disruption of this metabolism led to a decrease in the quantum yield.38,39 Nisin is a well-known pore-forming bacteriocin 40 active against Listeria, and other Gram-positive species were used in this study as the positive control in the bioluminescence inhibition assay. Interestingly, the treatment of E. coli cells by nisin showed dose-dependent progressing-in-time inhibition of bacterial luminescence. Generally, Gram-negative bacteria are not sensitive to the action of LAB-bacteriocins such as nisin, but Matsusaki et al. showed 41 that growth of E. coli was inhibited by a high concentration of nisin. So, it was demonstrated that LAB-bacteriocins under the relatively high concentration can be active against Gram-negative bacteria. Moreover, Kuwano et al. 42 revealed that nisin Z can make the E. coli cells permeable, but such effect was remarkably reduced in a salt environment. Indeed, it is known that the antimicrobial activity of bacteriocins can be affected by the ionic strength of the medium and various competitive proteins. Na+, Ca2+, and Mg2+ ions can inhibit the antimicrobial activity of cationic peptides by competing for binding sites on the cell surface, 33 as well as by modulating the secondary structures of peptides in aqueous solution. 43 The presence of salt in the medium did not affect the biological activity of the BLIS, which effectively inhibited the viability of strains of both L. monocytogenes and E. coli at various ionic strengths. The proteins present in the medium can also modulate antimicrobial activity. 44 These proteins can bind to small cationic peptides and reduce their effective concentration, thus inhibiting antimicrobial activity.45,46 The BLIS activity, which was estimated based on the ability to inhibit the growth of Listeria colonies, was not significantly reduced in the presence of high concentrations (10 mg/ml) of bovine serum albumin. At the same time, the antimicrobial effect of the BLIS against E. coli was dose dependent and was completely absent at the highest concentrations of bovine serum albumin.
Conclusion
In conclusion, we would like to note the following points. The first significant achievement of this study was the isolation of a novel BLIS from E. faecium ICIS 8, which was characterized by a unique primary peptide sequence. This BLIS exhibited high bactericidal activity against the L. monocytogenes by disrupting membrane integrity. In addition, this study described the interaction between the BLIS and Gram-negative E. coli K12 TG 1. In particular, the interaction between the BLIS and the bacterial surface was demonstrated by changes in the cell's zeta-potential, followed by the permeabilization of bacterial membranes, as determined using fluorescent spectroscopy and AFM. It was shown that peptide concentration is a key factor that determined the BLIS activity against Gram-negative E. coli K 12 TG 1.
Footnotes
Acknowledgments
This work was supported by a stipend of the President of Russian Federation for young scientists (Registration Nos. SP-943.2015.4 and SP-2093.2015.4): Russian Foundation for Basic Research (Project No. 16-34-50077) and by Fundamental Research Projects of Ural Branch of Russian Academy of Science (Project 15-4-4-28). The research was partially performed using an equipment of the Center of Shared Scientific Equipment “Persistence of microorganisms” of ICIS UB RAS. The authors thank Prof. Dmitry G. Deryabin (“State Research Center of Dermatovenereology and Cosmetology”) for some important remarks improving the article and Dr. Anna Tolmacheva and Dr. Ludmila Vlasenko (“Orenburg State University”) for bioluminescence assay technical assistance.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.