
Abstract
Shigella flexneri is one of the most prominent pathogenic bacteria in developing countries. In the battle against shigellosis and other bacterial diseases, antibiotic resistance has become an increasing global public health threat. Although the serious phenomenon of multidrug resistance (MDR) has been identified as one of the top three burdens on human health, resistance mechanisms are still poorly understood at the molecular level. In this study, we analyzed genomic data and the evolution of resistance in Shigella flexneri under sequential selection stress from three separate antibiotics: ciprofloxacin (CIP), ceftriaxone (CRO), and tetracycline. Through whole-genome sequencing, 82 chromosomal antibiotic resistance genes were identified. Re-sequencing of the evolved populations identified single nucleotide polymorphisms (SNPs) that contributed to MDR and SNPs that were specific to a single drug. A total of 40 SNPs in 8 genes and 3 intergenic regions, including mutations in metG (L582R) and 1538924, 1538924, and 1538924, appeared under each antibiotic. Several nonsynonymous mutations in gyrB (S464Y), ydgA (E378A), rob (R156H), and narX (K75E) were observed under selective pressure from CIP or CRO. Based on a bioinformatic analysis and previous reports, we discuss the contribution of these mutated genes to resistance. Therefore, more circumspect selection and use of antimicrobial drugs for treating shigellosis is necessary.
Introduction
S
The first identified Shigella species was S. dysenteriae, followed rapidly by the identification of S. flexneri at the end of the 19th century. Shigellosis became a notorious, widespread epidemic during World War 1 with the transmission of strain NCTC1, a 2a lineage of S. flexneri.3,4 S. flexneri has been identified in all areas of the world and has developed unprecedented diversity, with no fewer than 20 serotypes: 1a, 1b, 1c, 1d, 2a, 2b, 2v, 3a, 3b, 4a, 4av, 4b, 4c, 5a, 5b, X, Xv, Y, Yv, 6 and 7b,5,6 although serotype 2a is still highly prevalent.
To counter the blight caused by shigellosis, a large number of antibiotics have been used in clinical therapy, ultimately leading to widespread antibiotic-resistant bacteria. The systematic appearance of antibiotic resistance in pathogenic bacteria remains a global problem.7,8 One of the main reasons for the rapid accumulation of resistance in Shigella is likely the inappropriate and excessive use of antibiotics among outpatients in China. 9
Bacteria can achieve resistance via the sequential accumulation of multiple spontaneous mutations, the horizontal transfer of resistance genes, or changes in the expression of chromosomal genes.10–12 The evolution of resistance through access to single spontaneous mutations is particularly relevant for several types of antibiotics, such as quinolones, for which high levels of resistance can result from a single-locus mutation in genes in the quinolone resistance-determining region (QRDR) (gyrA, gyrB, parC, and parE).13,14 However, for most antibiotics, developing a high level of resistance requires multiple mutations. 15
Despite decades of research, our knowledge of the mechanisms underlying bacterial drug resistance is still incomplete. Therefore, extensive investigations are required to expand on and supplement our knowledge of antibiotic resistance mechanisms.
In this study, we not only analyzed the antibiotic resistance genes (ARGs) in the S. flexneri 2a 301 chromosome but also explored the undiscovered mechanisms underlying resistance under the continuous action of antibiotics. Through this multi-level study, we have assessed a considerable amount of genomic information on S. flexneri 301 and many other Shigella isolates reported over a long period, which will contribute toward understanding antibiotic resistance and provide guidance for addressing shigellosis.
Materials and Methods
Storage and antimicrobial susceptibility testing
There are many existing records of the collection of S. flexneri 2a, strain 301 because it was identified by the General Hospital of the People's Liberation Army (PLAGH) in 1984. In this study, S. flexneri 2a 301 isolated from bacillary dysentery patients decades ago and stored by the Chinese Center of Disease Control and Prevention (CDC) was used. The bacterium was grown in brain heart infusion broth (BD) for 24 hr under aerobic conditions and stored in 25% glycerol at −80°C.
We tested S. flexneri 301 for resistance against 25 modern antimicrobial drugs by using the microdilution method in 96-well plates, as recommended by the Clinical and Laboratory Standards Institute (CLSI). 16 The antibiotics (obtained from Sigma) included ampicillin (AMP), piperacillin (PIP), amoxicillin (AMX), cefazolin (CFZ), cefuroxime (CFX), cefoxitin (FOX), ceftriaxone (CRO), ceftazidime (CAZ), cefotaxime (CTX), cefepime (FEP), cefoperazone (CFP), imipenem (IPM), aztreonam (ATM), chloramphenicol (CHL), tetracycline (TC), erythromycin (E), rifampin (RIF), streptomycin (STR), gentamicin (GM), amikacin (AK), cotrimoxazole (COT), trimethoprim (TMP), norfloxacin (NOR), ciprofloxacin (CIP), and levofloxacin (LVX). The Escherichia coli ATCC25922 strain was used for quality control.
De novo sequencing and genome annotation
Genomic DNA from Shigella flexneri 301 strain (SF) for de novo sequencing was extracted with a DNA Isolation Kit (OMEGA) and sequenced on the HiSeq 2000 technology platform (Illumina, Inc., San Diego, CA) by using a paired-end strategy at Beijing Genomics Institute (BGI), Shenzhen, China. Automatic generation of qualified clusters and sequencing were performed on paired-end 2 × 90 bp reads. Clean data were acquired by filtering out adapters, duplications, and low-quality reads from the total data.
The assembly of short reads into genome sequences used NC_004337 (Sf.2a 301) as a reference genome and was performed with the SOAPdenovo17,18 assembler, with the key parameter Kmer value set to 71 for optimal results. The assembly results were then locally assembled and optimized according to paired-end and overlap relationships via mapping reads to contigs.
Functional annotation was accomplished by an analysis of protein sequences. We aligned genes with databases to obtain their corresponding annotations.19–26 In particular, we identified protein orthologs of ARGs by using the Antibiotic Resistance Genes Database (ARDB). 27 A gene was annotated as an ARG if the BLAST value was beyond the set threshold of amino acid sequence identity over 90%. To ensure biological meaning, the highest-quality result was chosen as the gene annotation and was completed by performing BLAST searches of the genes against each database.
Bioinformatics analysis
We compared the SF genome data assembled in this study with the reference genomes (complete and draft) of Shigella and E. coli from the National Center for Biotechnology Information (NCBI). A comparative genomics analysis was performed to explore the diversity of the relationships and the evolution of some Shigella and E. coli reference genomes. We used annotations to define core and pan genomes 28 and to establish a linear synteny analysis of the SF and reference genomes at the amino acid level to study the differences between each reference genome. 29 To perform a phylogenetic tree analysis, we generated a multiple sequence alignment (54 S. flexneri, 2 S. boydii, 2 S. sonnei, 1 S. dysenteriae, and 1 E. coli) by using TreeBeST. 30
Laboratory evolution experiment and re-sequencing
After determining minimum inhibitory concentrations (MICs), we conducted experiments to establish a series of drug-resistant strains. S. flexneri 301 was grown in Luria-Bertani (LB) medium (BD) that was supplemented with CIP, CRO, or TC at 0.25 × MIC at 37°C with shaking at 250 rpm until the culture reached an OD600 of ∼0.5. At this OD value, the bacteria had adapted to this fixed antibiotic concentration, and a higher drug concentration was necessary to maintain selection pressure to ensure that the population achieved a higher resistance level. Hence, the strain was successively cultured in medium containing twofold-increased concentrations to obtain 10 drug-resistant generations, in which the MICs had significantly improved. Under the same conditions, we also cultivated S. flexneri 301 in LB medium with no antibiotic as the control group.
The re-sequencing of nine antibiotic-induced strains was also performed on the HiSeq 2000, and the sequencing conditions were similar to those for the de novo sequencing, except that different key parameter Kmer values ranging from 23 to 81 were used for optimal results when they were assembled. The nine re-sequenced genomes were compared with SF to identify resistance-related single nucleotide polymorphisms (SNPs) in mutated genes. All the SNPs in mutated genes were tested in the control group by Sanger sequencing.
Results
Antibiotic-resistant phenotype
After being revived, the S. flexneri 301 isolate was tested to determine MICs by using 25 modern antimicrobial drugs. The results of the susceptibility testing suggested that the strain was sensitive to most of the antibiotics, but its resistance to several common drugs was still quite serious.
The resistance to TMP (64 μg/ml) was the highest, followed by AMP (32 μg/ml), COT (32 μg/ml), CHL (32 μg/ml), ATM (16 μg/ml), RIF (16 μg/ml), and E (8 μg/ml). The 301 strain exhibited resistance to PIP, AMX, and STR, with MICs ranging from 2 to 4 μg/ml. Fortunately, the 301 strain did not show resistance to any of the cephalosporin (CFZ, CFX, FOX, CRO, CAZ, CTX, FEP, CFP) or fluoroquinolone (CIP, NOR, LVX) antibiotics, with CRO (0.0625 μg/ml), CAZ (0.125 μg/ml), CTX (0.125 μg/ml), CIP (0.02 μg/ml), and NOR (0.16 μg/ml) all exhibiting MICs under 0.2 μg/ml. In addition, the strain was susceptible to the aminoglycosides STR, GM, and AK at a concentration of 1 μg/ml. Although the MIC for TC was 1 μg/ml, this is within the scope of inhibiting bacteria. The results showed that this strain possessed slight multidrug resistance (MDR).
Overview of de novo sequencing data
We sequenced the most epidemic S. flexneri strain (301) by using next-generation sequencing technology and constructed a high-quality complete genome (SF) (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/mdr).
Using the Illumina HiSeq 2000 sequencing platform, 732 MB of data were produced for the sampled SF. Based on the assembly results for the sampled SF, we present a chromosome sequence of 4,660,005 bp in length, with an average guanine-cytosine (GC) content of 50.87%; the number of scaffolds was 28, and the number of contigs was 95. Based on the genomic analysis of SF, we found that the genome contained 4,832 genes, and the total length of genes was 4,009,839 bp, accounting for 86.05% of the genome. The number of tandem repeat sequences was 78, and the total length of tandem repeat sequences was 11,024 bp, constituting 0.2366% of the genome.
Bioinformatic analysis
After capturing genomic information and genetic variation through Illumina sequencing, we compared the SF genome with some available Shigella and E. coli genomes.
A phylogenetic analysis of gene families was used to identify the evolutionary relationship of SF with other Shigella and E. coli strains that have diverged in different countries and regions. SF showed the closest evolutionary relationship with NC_004337, despite the presence of 51 InDels and 317 SNPs in SF. We used other Shigella species and E. coli genomes along with the completed or draft genomes of 53 S. flexneri serotyping strains available at NCBI to establish the phylogenetic relationships among a broader set of isolates (Fig. 1). Analysis of this phylogenetic tree showed that S. flexneri serotypes 1–5 and X, Xv, and Y were virtually monophyletic within Shigella and Shigella spp. originating in the Escherichia genus, as previously reported.31,32 However, we also found that the evolution of S. flexneri was characterized by national independence.

Phylogenetic analysis of the Shigella flexneri 301 strain genome and reference genomes. Our phylogenetic tree shows the evolutionary relationships among reference genomes available at NCBI. These strains were isolated in different countries and regions. Red circle, China; purple circle, France; yellow circle, Bangladesh; blue circle, the United States; green circle, Japan. SF, Shigella flexneri 301 strain.
To investigate similarities with other Shigella and E. coli K12 genomes, we compared their basic information and genome features with those of SF. Although the years and areas of these strains were different, they had a similar GC%, ranging from 50.60% to 51.10% (SF 50.87%). However, there was a large difference in SNPs and coverage between SF with the S. flexneri genome and SF with other Shigella and E. coli K12 genomes (Table 1).
Coverage: similarity between SF and the reference genome.
GC%, guanine-cytosine content; SF, Shigella flexneri 301 strain; SNP, single nucleotide polymorphism.
The genome of SF was similar in amino acid linear synteny to that of S. flexneri, especially NCTC1 (only an inversion), but there were great differences compared with other Shigella strains and E. coli K12 (Fig. 2 and Supplementary Fig. S2). They shared the most (4,455) core genes and a high degree (99.68%) of coverage, but with the least (459) SNPs compared with the S. flexneri 2a NCTC1 (Table 1). However, although there was a large number (3,852 to 4,109) of core genes and a high similarity (84.58% to 87.18%) of coverage, the number of SNPs was tremendous compared with SF with Sd197 (65,674), Sb227 (47,651), and Ss046 (47,596). Unexpectedly, although there were 57,950 SNPs in E. coli K12 compared with SF, they shared 4,091 core genes, 82.58% genome similarity, and good amino acid linear synteny (Table 1 and Supplementary Fig. S2).
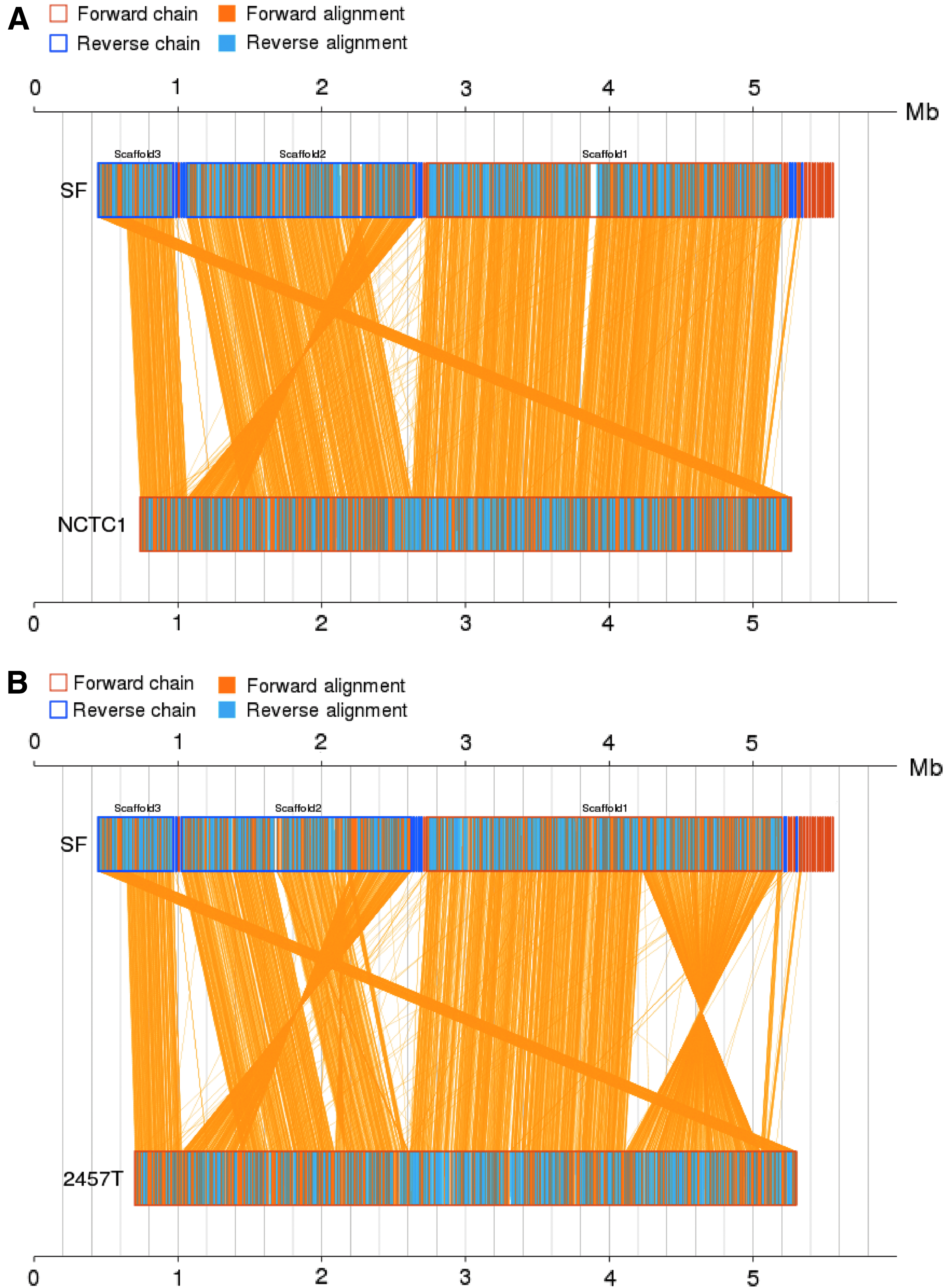
Linear synteny analysis of SF and S. flexneri 2a reference genomes at the amino acid level. From top to bottom; the first axis is the first reference sequence; the second and third axes are the reference sequences; and the target sequence is located between the axes. The yellow box represents the forward chain, and the blue box represents the reverse chain within the axis region. In the box on the axes, the yellow region represents the amino acid sequence on the forward chain of this genome sequence, and the blue region represents the amino acid sequence on the reverse chain of this genome sequence.
Functional gene annotation
After completion of the analyses described earlier, we annotated the 4,832 genes by using 10 databases that are commonly employed in bioinformatics. Functional annotation was completed through BLAST searches of the genes against different databases, and we ultimately obtained abundant gene annotation information (Table 2). According to an analysis of the annotation results, we identified 82 ARGs, 52 of which encoded MDR efflux pumps (Table 3). This high number of efflux pump genes was organized into five different families: the major facilitator superfamily (MFS), the multidrug and toxic compound extrusion family (MATE), the resistance nodulation cell division family (RND), the small multidrug resistance family (SMR), and the ATP-binding cassette family (ABC).33,34
PBP penicillin-binding protein.
HTH (helix-turn-helix) marR-type DNA-binding domain.
drug/metabolite transporter (dmt) superfamily.
MarC multiple antibiotic resistance protein.
HTH (helix-turn-helix) araC-type DNA-binding domain.
ATP, Adenosine triphosphate; MDR, multidrug resistance; MFS, major facilitator superfamily; QRDR, quinolone resistance-determining region.
Genes encoding various types of enzymes related to drug resistance, such as the Bl1_ec gene, encoding the enzyme ampC, which can hydrolyze beta (β)-lactam antibiotics, and QRDE genes (DNA gyrase: gyrA, gyrB and DNA topoisomerase: parC, parE) were present in SF, as well as other enzymes mediating bacterial drug resistance in different indirect ways. We also found 10 penicillin-binding proteins, which contribute to β-lactam resistance.
Among the examined databases, ARDB is a special database containing a vast array of ARGs. Using this database, we identified 20 ARGs encoding enzymes and MDR efflux pumps (Fig. 3). Among these pumps, three efflux pump systems have been reported to interact with tolC, an outer-membrane channel protein, to form tripartite transperiplasmic complexes: macAB-tolC, mdtEF-tolC, and acrAB-tolC, which are responsible for the direct extrusion of aminoglycosides, glycylcyclines, macrolides, β-lactams, fluoroquinolones, and acriflavine from the cell.

SNPs and ARGs identified in this study. This figure mainly includes three groups: (1) The red region at the top represents the SNPs identified by re-sequencing. The rectangles and polygons represent nonsynonymous and synonymous mutations, respectively, and the dotted line represents mutated loci in noncoding regions of genes. Different arrows indicate the mutation caused by each antibiotic. (2) QRDE consists of four resistance genes. DNA gyrase subunit B (gyrB) also exhibits a nonsynonymous mutation under the pressure of ciprofloxacin. (3) The chromosomal ARGs are annotated in the ARDB database. ARGs, antibiotic resistance genes; SNP, single nucleotide polymorphism.
Next, we established a statistical map for the ARGs of all reference genomes (Fig. 4) and compared the ARGs that were consistent between SF and other phenotypes. Interestingly, the efflux pump genes acrAB-tolC, metH, mdtK, and mdtL existed in all the reference strains, and all the S. flexneri lineages exhibited the bcr gene (except for M90T) but lacked mdtG and mdtM. In addition, there was a significant β-lactamase (bl1_ec) conferring resistance to penicillin and cephalosporin. This enzyme can break open the β-lactam antibiotic ring and deactivate the molecule's antibacterial properties, and it is widely conserved throughout most of Shigella. Some special genes (dfra5, sul1, ctx-m, qnrS, dfra14, tem1, and tetA) detected in only one or two strains are capable of conferring resistance to specific antibiotics.

Statistical chart of ARG orthologs in reference sequences from Shigella and Escherichia coli from the ARDB database. Red and green represent the presence or absence of the gene in the corresponding genome, respectively.
The chromosomal ARG complements of these strains identified by using ARDB were similar, whereas the ARGs present on plasmids showed large differences.
Evolution and re-sequencing of induced drug-resistant strains
According to the results of susceptibility testing, we selected three different types of antibiotics (CIP, CRO, and TC) to select directional resistance in S. flexneri 301. The laboratory evolution experiments accumulated ample data related to the genetic changes underlying the multiple resistance phenotypes. Over time, the resistance level increased dramatically, and 30 strains acquired various resistance levels, ranging from sensitive to highly resistant. At the end of the evolution experiments, the MIC values for CIP, CRO, and TC had become 64, 64, and 128 μg/ml, respectively (increases of 3,200-, 1,024-, and 128-fold, respectively).
A comparison of the patterns of evolution revealed that resistance to the three antibiotics increased gradually and consistently in the first five generations, then increased in a stepwise fashion in the generations of CIP5-7, CIP8-9, CRO8-10, and TC6-7, 8-9. Following the characteristics of the evolving dose-response curves, we selected CIP- and TC-induced strain generations 5, 7, and 10 and CRO-induced generations 5, 9, and 10 for re-sequencing (Fig. 5).

Gradient antibiotic stress induces high levels of drug resistance in spontaneous adaptive trajectories. The relationship between the incubation period and MICs is shown. The horizontal coordinates were the number of generations under the action of antibiotics. Strains from the successive evolution experiment were grown in 10 gradient concentrations of the antibiotics CIP, CRO, and TC, and the corresponding MICs were measured. Based on the characteristics of this line graph, the generations indicated by filled points were selected for re-sequencing. CIP, ciprofloxacin; CRO, ceftriaxone; MICs, minimum inhibitory concentrations; TC, tetracycline.
Whole-genome re-sequencing of the nine evolved populations identified mutations that were either specific for resistance to a particular drug or shared in resistance to multiple drugs. Through Illumina whole-genome re-sequencing, we obtained a vast amount of high-quality genetic information, with the only exception being CRO-9, which showed a low average sequencing depth (Supplementary Table S1). All sequences have been submitted to NCBI Sequence Read Archive (SRA), and the accession number is SRP106446.
Subsequently, we identified a list of SNPs by comparing the re-sequenced genomes with SF and verified them through Sanger sequencing, with most of the SNPs being confirmed (Fig. 3, Supplementary Fig. S1, and Table 4). Each sequenced strain exhibited two or more SNPs, but we reliably identified only one SNP in each gene.
Each mutation was found by Illumina sequencing and verified by Sanger sequencing.
Table 4 shows all 40 verified SNPs, located in eight genes and three intergenic regions. The majority of these gene mutations resulted in nonsynonymous amino acid substitutions (15 of 40) in five genes and synonymous substitutions (7 of 40) in two genes; the other SNPs were located in the ppiA gene promoter (18 of 40). Among these mutated genes, only gyrB of CIP-5, 7, and 10 was directly related to ciprofloxacin resistance. Notably, the same mutations in metG and the promoter of ppiA were found to have occurred in each antibiotic-induced population. The mutated genes rob (CIP-10) and narX (CRO-10) are components of the mar-sox-rob regulon activator and the narX-narL two-component regulatory system, respectively.
Discussion
Shigella is one of the major etiologic causes of shigellosis worldwide. There are currently no specific therapies available for this disease, and good hygiene is the best precautionary measure against bacillary dysentery. 35 Despite improvements in public sanitation, outbreaks of shigellosis are still quite serious.36,37
Due to the lack of an available vaccine, antimicrobial drugs have been extensively used for the treatment of shigellosis. However, the MDR of Shigella means that treatment is not straightforward, although some regional variation in resistance exists. 35 Mather confirmed that resistance existed in S. flexneri before the appearance and widespread use of antibiotics, and S. flexneri is already a well-adapted pathogen and continues to respond to selection pressures. 38 Although shigellosis is still intractable due to its epidemiological distribution and wide-ranging antibiotic resistance, whole-genome sequencing might provide information that could help researchers develop an effective treatment scheme.
Through a bioinformatic analysis, we studied S. flexneri strains ranging from the earliest isolate, NCTC1, to the newly isolated X and Y serotypes, in addition to analyzing E. coli and other species of Shigella. Our phylogenetic analysis and linear synteny showed that the genome of Shigella presents high homology and has been relatively stable during the process of evolution. The 301 chromosome is very similar to those of other S. flexneri strains and shares more than 90% of its core genes with reference genomes. In addition, our study suggested that the 301 strain may have evolved directly from NCTC1 and that these strains share a common ancestor in E. coli, as previously reported. 35
Intrinsic ARGs are also highly conserved and could be responsible for the persistent inheritance of antimicrobial resistance in Shigella. 4 Most currently used antibiotics are produced from environmental microorganisms or their derivatives. 39 Therefore, the original resistance genes in environmental bacteria had different ecological purposes, such as detoxification, signal trafficking, or metabolic functions.40,41
The intrinsic ARGs in the 301 chromosome are similar to other chromosomal ARGs of Shigella, most of which existed before the widespread clinical use of antibiotics and the development of MDR. However, the ARGs present on plasmids show clear differences in each reference strain and usually mediate resistance to a single drug or class of drugs. Compared with chromosomal ARGs, plasmid ARGs may cause greater harm because of their transferability and rapid spread from one strain to another, leading to the dissemination of resistance. 42
In this study, a panel of chromosomes were discovered in the 301 strain. These ARGs encode enzymes, efflux pumps, and transcriptional regulation factors, conferring the potential ability to resist most types of antimicrobial drugs. Although the 301 strain is sensitive to most modern antibiotics, resistance is likely to speed the development of adaptation to evolutionary pressure.
Accumulation of mutations in genes encoding transporters, targets, and proteins could activate conferred resistance. 41 Our evolution experiments generated important mutations related to drug resistance that allowed us to clarify potential resistance mechanisms. Through a comparison of the mutation patterns associated with the three antibiotics, we found that the bacteria exhibited different measures to cope with sequentially increasing selective pressure. For each of the populations, the L582R mutation in metG, which is located in the tRNA-binding region, was ubiquitous.
MetG encodes methionyl-tRNA synthetase (MetRS), an enzyme essential for viability that functions to covalently link methionine amino acids to their cognate tRNA molecules. 43 MetRS is required not only for elongation in protein synthesis but also for the initiation of all mRNA translation through initiator tRNA (fMet) aminoacylation. A mutation in methionyl-tRNA synthetase (MetG) might result in reduced growth rates relative to the wild type, which would affect sensitivity and susceptibility to antibiotics.43,44 The bacterial metG gene, or MetRS, is an unsurpassed target that presents unlimited potential for the development of antibacterial agents. Topical research is mainly concentrated on MetRS inhibitors as late-model, effective antibiotics.
Regarding selection pressure from the first-line drug for treating shigellosis, CIP, resistance occurs mainly through mutations in the QRDRs of gyrA and gyrB, encoding DNA gyrase, and parC and parE, encoding topoisomerase. Previous studies have shown that most quinolone resistance in Shigella is mediated by mutations in gyrA (Ser83Leu, Asp87Gly/Asn, or His211Tyr) and parC (Ser80Ile).45,46 Although the Ser464 mutation in gyrB has been described as being associated with quinolone resistance,47,48 its prevalence in clinical isolates is very low, and it was discovered in Shigella for the first time.
With the increasing use of CIP, the new mutation R156H in rob (CIP-10) has developed to cope with the pressure to survive. A previous study on the evolution of resistance by Toprak et al. 15 revealed another mutation in rob, E262K, that caused a high level of resistance to CHL. The right origin-binding protein rob, which belongs to the AraC/XlyS family, is a component of the so-called marbox (MarA-SoxS-rob) global transcription regulator of MDR. 49 Many bacteria possess a rob gene that responds to drugs by activating the expression of genes encoding proteins that are involved in antibiotic efflux and detoxification through interacting as a monomer with the highly degenerate sequence recognized by MarA and SoxS.
Many multidrug efflux systems, such as the AcrAB-TolC complex, play an important role in the intrinsic resistance of bacteria against antimicrobial compounds and are regulated by marbox. 50 A great deal of evidence shows that rob is involved in the regulation of genes conferring an MDR phenotype in various species of the Enterobacteriaceae family and promotes resistance to antibiotics through upregulation of the multidrug efflux pump AcrAB-TolC.50,51 Another principle role of rob is to mediate MDR by upregulating the expression of micF antisense mRNA, a small inhibitory RNA that downregulates the outer-membrane porin OmpF to limit the uptake of antibiotics such as fluoroquinolones and β-lactams.52–54
Similarly, two nonsynonymous mutations found in populations that evolved under CRO inhibition emerged in metG and another regulatory system gene, narX. However, regarding the mechanism underlying the inhibition of bacterial cell wall synthesis, β-lactam antibiotics are bactericidal drugs. The reduction in the growth rate caused by metG mutations may lead to increased function under the threat of CRO. Therefore, the MICs increase slowly and continuously before CRO-8.
Under the pressure of high concentrations of CRO, one member of the two-component system (TCS) of signal transduction, narX, developed a mutation (K75E). Bacterial species have a wide range of TCSs to control numerous physiological behaviors, such as virulence, environment stress responses, biofilm formation, central metabolism, and antibiotic resistance.55,56
TCSs play essential roles in prokaryotic organisms to monitor and rapidly deploy physical responses to meet changing environmental conditions. As a typical TCS, the narX-narL regulator pair consists of two signal transducers. The sensor kinase narX senses certain environmental changes and accordingly modulates the phosphorylation state of the response regulator. The response regulator narL controls the level of gene expression to perform related physiological behaviors.56,57 Although the mechanism of resistance mediated by narX is unclear, previous research showed that this TCS can confer increased β-lactam antibiotic resistance through the overexpression of the response regulator narL. 58
Under pressure from TC, only one nonsynonymous mutation appeared. However, it definitely did not represent a separate mechanism of resistance. Intrinsic MDR ARGs may play an important role in resistance.
We cannot ignore the indispensable gene peptidyl-prolyl cis-trans isomerases (ppiA) identified in association with each of the tested drugs. This enzyme has mainly been studied in relation to cancer but also shows crucial functions in bacteria. It facilitates proper protein folding by increasing the rate of transition of proline residues between the cis and trans states, which is a rate-limiting step in protein folding. 59 Although ppiA is not as essential as surA for viability, it influences the growth rate and has substantial effects on antibiotic sensitivity. 60
In this study, in addition to identifying major resistance potential through whole-genome sequencing, we analyzed the coping mechanisms under each tested antibiotic selective pressure, as previously reported. Regardless of whatever effect is conferred by mutations, these genes contribute to the tolerance of strains to antibiotic threats in various manners. However, this is just the tip of the iceberg regarding bacterial drug resistance mechanisms. With their enormous ability to select resistance to antimicrobial drug stress, bacteria become resistant to all types of antibiotics. Therefore, we can turn our attention to the development of functional gene inhibitors for decreasing the accumulation of resistance ability.
Overall, multiple antibiotic resistance in bacteria has become an inevitable problem that is still poorly understood at the molecular level. Widespread drug resistance is a complex process, involving various types of resistance genes, regulatory mechanisms, and quorum-sensing synergies, resulting in high-level resistance ability. Continued efforts to study the underlying resistance mechanisms, achieve the appropriate use of antibiotics, and develop novel antimicrobials or inhibitors of bacterial survival and metabolic activity are needed to improve treatment strategies and to ameliorate worldwide resistance against frequently used antibiotics.
Footnotes
Acknowledgments
This work was supported by funds from the National Natural Science Foundation of China (31272603, 31101836). The authors are grateful to the Chinese Center for Disease Control and Prevention for providing the bacteria for this study.
Authors' Contributions
Conceived and designed the experiments: J.Y.Z. and Z.Z. Performed the experiments: Z.Z. Analyzed the data: Z.Z., B.L., and X.Z.Z. Contributed reagents/materials/analysis tools: F.S.C. and J.R.N. Wrote the article: Z.Z.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.