
Abstract
gyrA is a clinically validated therapeutic drug target that catalyzes changes in DNA topology by introducing negative supercoils into DNA. The broad-spectrum quinolones are hitherto the most potent antibacterial agents targeted toward gyrA. Increasing resistance to quinolones is accompanied by several mutations in the quinolone resistance-determining region of gyrA. In the present study, we report a comprehensive picture of the dynamic behavior of wild-type, T86I, and T86I/P104S mutants (MTs) of gyrA from Campylobacter jejuni in complex with ciprofloxacin to unravel the atomistic details of the mechanism underlying resistance to quinolones. Our simulation results reveal that no substantial conformational changes were observed. It was observed that these mutations disrupted residue interaction network landscape to a significant extent, which would affect ligand binding affinity. A distinctive pattern of dominant motions was clearly discernible in wild and MT gyrA forms. The results reported in this study associate gyrA mutations with quinolone resistance and would pave a way for facilitating wet laboratory researchers to develop gyrA MT-based therapeutic strategies against resistant pathogens.
Introduction
S
Campylobacteriosis, the most common form of enteric bacterial infections, is usually self-limited, but in the persistent form, it requires antibiotic treatment with erythromycin and fluoroquinolones. Growing body of evidences has documented resistance to different antibiotics such as tetracycline, kanamycin, chloramphenicol, erythromycin, and ciprofloxacin in C. jejuni strains.6–8 Campylobacter resistance to ciprofloxacin, nalidixic acid, and tetracycline in the United States is concerning as evidenced by an alarming level of increased resistance to these antibiotics. Ciprofloxacin resistance in C. jejuni doubled from 13% in 1997 to 26% in 2011. 9
Fluoroquinolones are broad-spectrum antibiotics with improved pharmacokinetic profiles that inhibit DNA gyrase and topoisomerase IV by inhibiting relaxation of the supercoiled DNA, promoting breakage of double-stranded DNA and blocking progress of the DNA replication enzyme complex, which ultimately leads to damage to bacterial DNA and bacterial cell death.10,11 Various studies on clinical isolates of C. jejuni have reported mutations in the chromosomal genes that encode the subunits of DNA gyrase, which lead to increased minimum inhibitory concentration (MIC) values of fluoroquinolones.6,12–14 This observation suggests that the drug resistance may be related to specific interactions between the antibiotic and the target.
Although resistance to quinolone class is chromosomal in origin, other mechanisms such as alteration in drug permeability, active efflux pumps, and plasmid-mediated resistance mechanism have also been established as responsible for bacterial resistance to quinolones.6,15 The emergence of mutations, though, is the major underlying force that drives evolution. As these mutations lead to structural alterations and thus reduce binding affinity, consequently studying mutational resistance provides an invaluable tool in understanding bacterial genetics and resistance to particular antibiotics 16 and to eventually gain a holistic view of the related molecular changes.
In the present study, we report molecular dynamics (MD) simulations of C. jejuni wild-type (WT) gyrA and ciprofloxacin-resistant mutants (MTs) T86I (M1) and a double MT T86I/P104S (M2) in complex with ciprofloxacin to identify the molecular events underlying ciprofloxacin affinity differences reflecting different phenotypes, which ultimately lead to ciprofloxacin resistance.
Methodology
Preparation of starting structures
The full-length canonical structure of gyrA is encoded by a polypeptide of 875 amino acids with a molecular weight of about 97 kDa. The structure of gyrA from C. jejuni strain NCTC 11168 using Escherichia coli gyrA (PDB code: 1AB4) as template was homology modeled in Discovery Studio, version 4.1 (DS). Heteroatoms, crystallographic waters, and cofactor were removed from the structure before docking. In DS, the starting structures of the MTs were modeled in silico by mutating the target residues with desired amino acids keeping the secondary structure intact. The complex structures were generated by computationally docking the WT and MTs of gyrA with ciprofloxacin in DS. Initial constructed models were further energy optimized and used as starting structures for MD simulations.
MD simulations
The molecular system was simulated using MD in Amber11.0 17 and Ambertools1.5. Hydrogen atoms were added using the tLeap program implemented in Amber tools and parameters were assigned according to AMBER FF99SB force field. 18 The system was solvated with TIP3P water model 19 in an octahedral box with 15 Å buffer around the complex. Twelve Na+ counterions were added to maintain neutrality of the systems.
Each molecular system, that is, (1) native modeled complex of 1AB4, (2) modeled complex of 1AB4 with substitution of Thr to Ile at position 86, and (3) with substitution of Thr to Ile at position 86 and substitution of Pro to Ser at position 104, was energy minimized through three rounds of energy minimization of 1,000 steps each. In each set of minimization, protein was energy minimized for 500 steps of steepest descent, followed by 500 steps of conjugate gradient with a time step of 2 fsec to eliminate close van der Waals contacts. In the first and second minimization round positions, restraints of 10 and 2 kcal−1Å−2, respectively, were imposed, while in the third minimization, the entire system was minimized without any restraints.
After minimization, each system was then gradually heated from 0 to 300 K, followed by constant temperature equilibration at 300 K and 1 atm pressure for 100 psec. Following stabilization of thermodynamic properties, a 10-nsec MD simulation was performed for each system with an integration step of 2 fsec in an isothermal isobaric ensemble (Normal Pressure and Temperature, T = 300 K and P = 1 atm) with periodic boundary conditions. NPT conditions were maintained with Langevin thermostat 20 and Berendsen barostat. 21 The SHAKE algorithm 22 was applied to constrain all covalent bonds involving hydrogen atoms.
Long-range electrostatic forces were treated using the particle mesh Ewald method 23 with a charge grid spacing of ∼1.0 Å and the charge grid interpolation on a cubic grid by setting the direct sum tolerance to 4.0 × 10−6. Short-range electrostatics and van der Waals interactions were evaluated using a 9.0 Å atom-based cutoff. Trajectory coordinates were saved every 2 psec for further analysis. After performing MD simulations, the root mean square deviations (RMSDs) and root mean square fluctuations (RMSFs) of the MD trajectories were analyzed with respect to each initial set of coordinates.
MD simulation protocol has been validated based on the modeled structure of WT-gyrA. The convergence of simulations was analyzed by monitoring parameters such as the energy components, RMSDs from the initial modeled structure, and RMSFs for WT-gyrA and each of the MTs (T86I, T86I/P104S).
Molecular mechanics Poisson–Boltzmann surface area calculations
Free energies were calculated using the conventional molecular mechanics (MM)-Poisson–Boltzmann surface area (PBSA) and MM-generalized born (GBSA) approaches in AMBER 11. Free energies were estimated by averaging the configurations that were extracted from a total of 2,500 frames at every 20 psec giving a total of 125 frames for energetic analysis during the last 5 nsec of MD simulation. The binding free energy profiles were calculated for WT and each of the MTs (T86I, T86I/P104S) using the following equations:
where T and S correspond to the temperature and the total solute entropy, respectively; Egas signifies gas-phase energy, which is the sum of internal energy (Eint), electrostatic (Eele), and van der Waals contributions (Evdw). Egas is evaluated using parameters from the FF99SB force field terms. The solvation free energy (Gsol) can be further decomposed into polar and nonpolar solvation states. The polar solvation contribution (GGB and GPB) is determined by solving PB and GB equations. The nonpolar solvation contribution (GSA) is estimated using 0.0072 kcal mol−1 Å−2 as the value for constant γ, and the solvent accessible surface area (SASA) is determined using a water probe radius of 1.4 Å. Dielectric constant values for solute and solvent were set to 1 and 80, respectively.
To determine the contribution of individual amino acids toward total binding free energy between ciprofloxacin and the WT/MT of gyrA, a per-residue decomposition analysis of the interaction energy for each residue was carried out using the GB model implemented in Amber11. Decomposition analysis was performed on l25 frames obtained during last 5 nsec of MD simulation. Amino acids contributing binding free energy more than 0.5 kcal mol−1 were identified as the hotspot amino acids. These hotspot amino acids add up most to complex stability and are crucial for the gyrA-ciprofloxacin interaction.
Principal component analysis
To investigate the direction and amplitude of the dominant motions of MD trajectories, essential dynamics (ED) calculations were performed using the principal component analysis (PCA) method by reducing the dimensionality of MD simulation data. PCA can help in identifying an essential subspace that probes pronounced motions corresponding to large-scale vibrational modes of groups of atoms in normal mode analysis. 24 Before analysis, the overall translational and rotational motions were excluded by translating MD trajectory to the geometrical center of the molecule and superimposing onto a reference structure. 25
PCA constructs the configurational space by orthogonal linear transformation in a new coordinate system to generate a covariance matrix (C). Associated eigenvectors (Vi), namely principal components (PCs), and eigenvalues (λi) are generated by diagonalization of the covariance matrix. Vi give a vectorial description of each component of the motion by indicating the direction of the motion, and λi represent the amplitude of eigenvectors along the multidimensional space. Projection Proj[M,PCi] of any frame M onto
i
th PC is calculated by Equation (6):
where Mα is the Cα atom of every structure after overlaying M with the reference structure. Projection highlights the time-dependent motions that the components perform in the particular vibrational mode. PCA was carried out using the PCAsuite software (http://mmb.pcb.ub.edu/software/pcasuite/) on (i) WT, (ii) T86I, and (iii) T86I/P104S MT models. MD trajectories of corresponding atoms were extracted using the PTRAJ program and analysis carried out for the last 5 nsec using 63 frames.
Residue interaction network analysis
The average structure derived from the last 5-nsec trajectory of each system, wild and T86I and T86I/P104S MTs, was used for constructing the residue interaction networks (RINs). Reduce program 26 adds hydrogen atoms to the input structure using local geometry. PROBE 27 identifies interacting amino acids by evaluating their atomic packing using small-probe contact dot surfaces. PROBE uses several scoring functions to quantify noncovalent interactions such as interatomic contact, hydrogen bonds, salt bridges, and pi–pi interaction. RINs generated from the averaged MD structures were visualized using RINalyzer. 28 Analyzing different RINs helps us to extract changes in the networks that reflect alterations in physicochemical characteristics of the structures.
Results and Discussion
Structural stability characterization
We carried out MD simulations of complexes of WT and MT gyrA from C. jejuni with ciprofloxacin to better understand the mechanism underlying gyrA resistance to fluoroquinolones at the atomic level. WT and MT gyrA (T86I and T86I/P104S) docked against ciprofloxacin were computationally simulated to complement experimental findings of increased MICs reported in clinical isolates harboring these mutations. To evaluate the conformational flexibility of the WT and MT structures, time-dependent RMSDs of the backbone atoms relative to the corresponding energy-minimized structure were monitored. RMSDs of WT and MT complexes estimated relative to the reference structure are plotted in Supplementary Fig. S1 (Supplementary Data are available online at www.liebertpub.com/mdr) as a function of time.
RMSDs were calculated from the last 5 nsec of the 10-nsec MD simulations. It was observed that all the molecular systems stabilized over the 10-nsec time course of simulation, with moderately small fluctuations in the models arising at the beginning of every production run. For all the three systems, the final RMSD values relative to the reference energy-minimized structure range from 1.3 to 1.8 Å. RMSDs in the native and MT forms of gyrA were relatively constant, which is mirrored by a narrow range of time-averaged RMSD values. It suggests that these mutations do not disrupt the backbone conformation or cause significant global change. It is speculated that due to these mutations, changes in the dynamic behavior of gyrA-ciprofloxacin complexes lead to reduced binding affinity.
RMSFs of each residue in WT and T86I and T86I/P104S MTs of gyrA are plotted in Supplementary Fig. S2 to monitor the effect of these mutations on dynamic behavior of complexes. RMSFs were computed relative to the average structures generated during the last 5 nsec of the 10-nsec MD simulations. The apex points of RMSFs for N-terminal residues and for residue ranges 250–260 and 370–450 were of different magnitude for WT and both MT trajectories, which imply that despite the fact that no considerable distortion was observed in the backbone structure, residue flexibilities in MT forms show large deviations (changed RMSF values) from those observed in WT (Supplementary Fig. S2).
The deviations in residue fluctuations may influence the interaction with ligand and thus the ligand binding efficiency. The regions of high RMSFs were not confined to the vicinity of mutated residues only, but were localized and distributed across the entire protein backbone, which suggests that the mutation effect is not restricted to its close vicinity, but it affects the entire protein structure. At some points, RMSF difference between WT and MTs was significantly higher (Supplementary Fig. S2), which further strengthens our argument that mutations introduced in the native structure impact the dynamic behavior of the protein. Residue flexibility was observed to be lowest in T86I/P104S, which would ultimately lead to reduced binding affinity.
Secondary structure content
The ensemble-averaged secondary structure populations of each amino acid residue for all three molecular systems are plotted in Fig. 1. Although WT and MT variant conformations appear to be dominated by α-helices, yet turns and antiparallel β-sheet structures were apparently visible. Other regular secondary structures were present in very small fractions. Pi-helix population was almost negligible for all the systems investigated and was almost absent from T86I MT. The helical content was radically higher than the β-sheet and turn contents in all molecular systems. The proportional contribution of various secondary structure types was similar across all three molecular systems and thus no significant changes were observed in the secondary structural content in ensemble-averaged structures of MTs from WT.
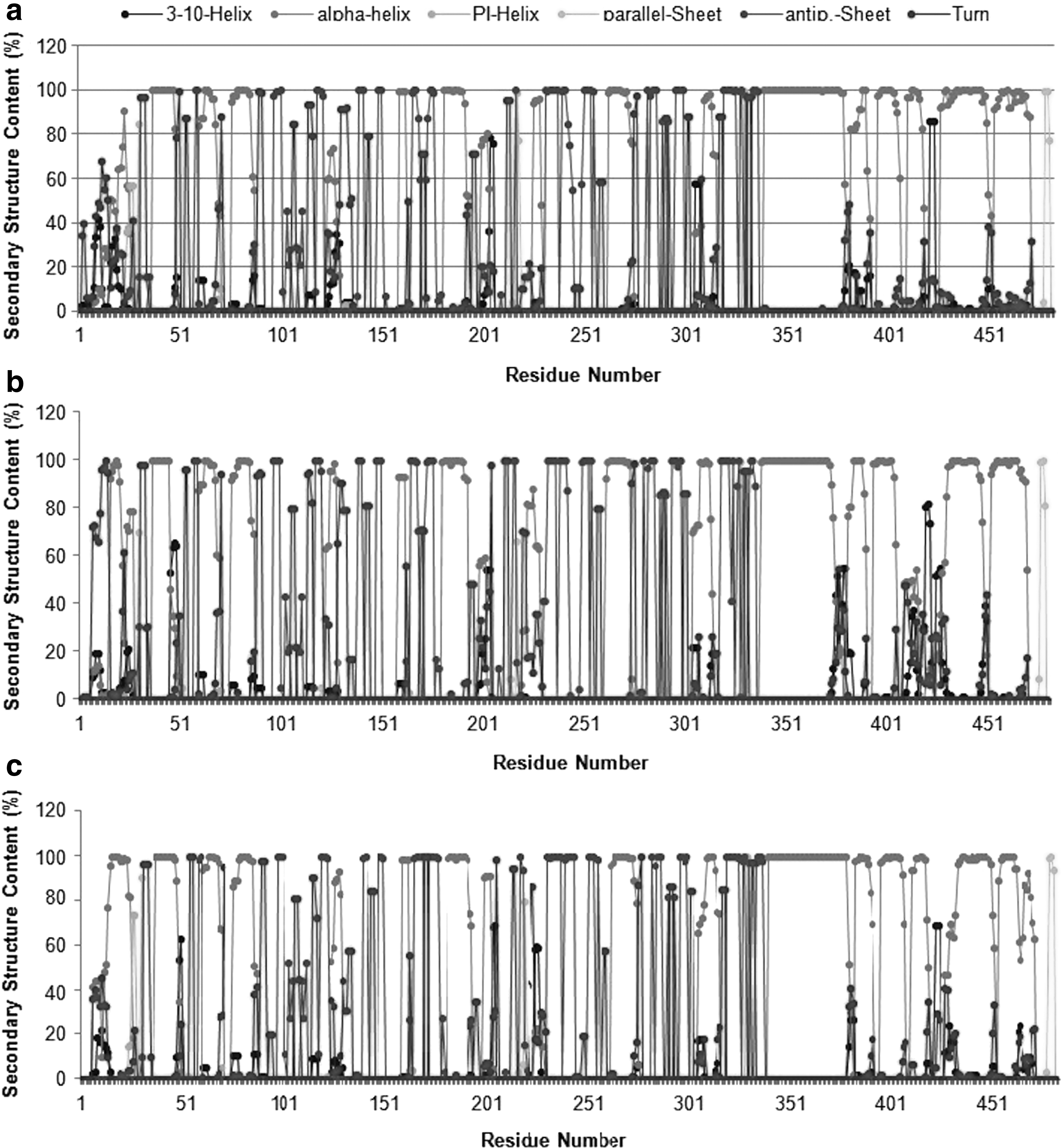
Average secondary structure contents of protein backbone residues in
These findings again point to the argument that the backbone conformations were not significantly altered due to mutations harbored in these structures. To investigate the dynamic structural changes of mutated residues and overall conformation of molecular complexes, we visualized MD trajectories using visual molecular dynamics. 29 It was observed that conformation of mutated residues as well as overall backbone conformation fluctuates in WT, T86I, and T86I/P104S MTs during the course of simulation. In Fig. 2, three snapshots saved for each molecular system at 0, 5, and 10 nsec of the MD trajectories reflecting conformational changes are shown.
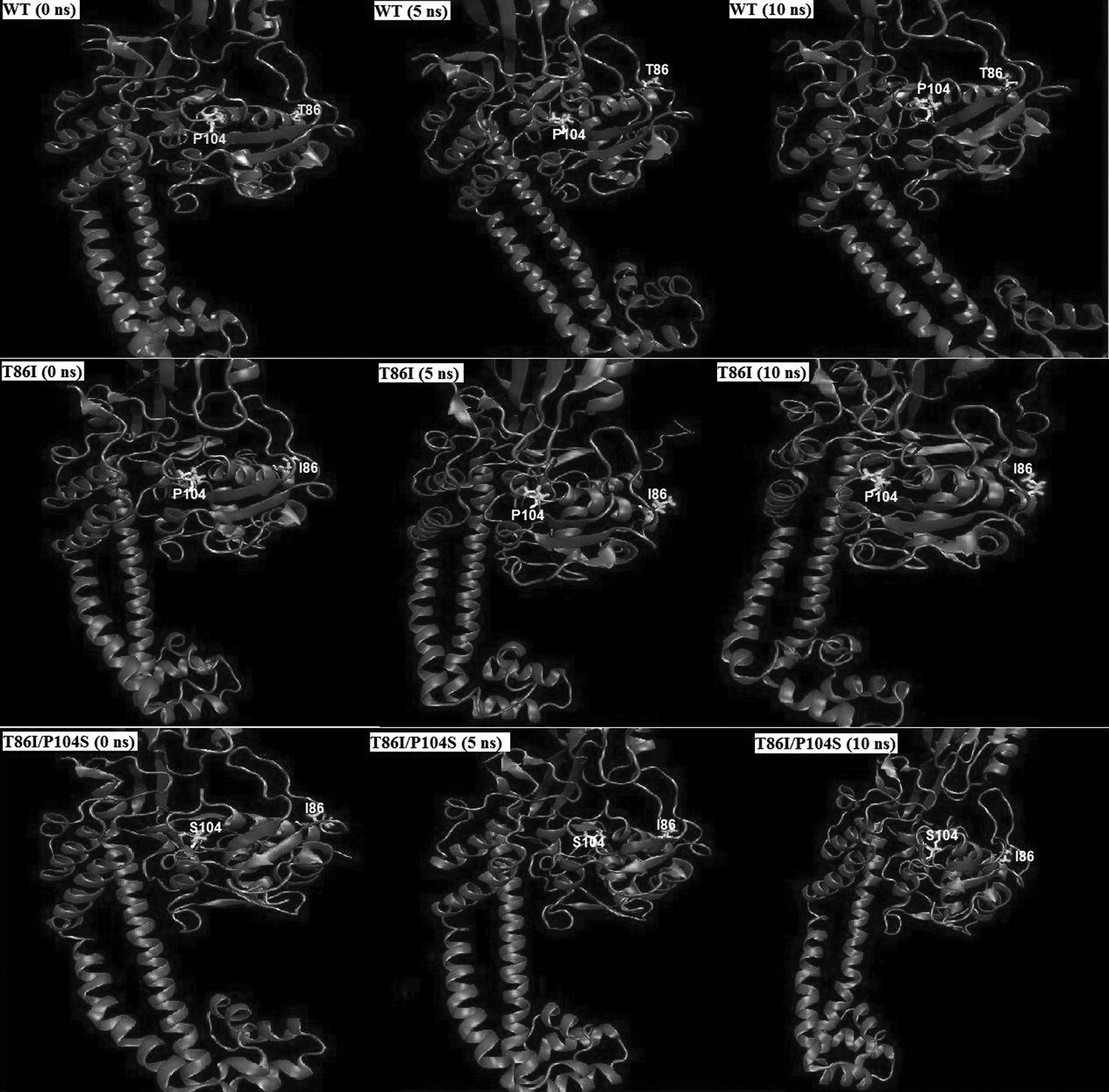
Observed conformational changes of the protein backbone and mutated residue positions 86 and 104 during the course of MD simulation in WT (Row 1), T86I MT (Row 2), and T86I/P104S MT (Row 3) at 0 nsec (start of production phase), 5, and 10 nsec (end of production phase). Conformational rearrangements of mutated residues as well as average ensemble structure consequently disrupt the active site landscape affecting ligand binding affinity. MD, molecular dynamics.
Several mutations in the quinolone resistance-determining region (QRDR) of the structural target gene, that is, gyrA, are associated with increased quinolone resistance in vivo, which accounts for reduced affinity toward quinolones. The QRDR region is mapped to residues 67–106 of E. coli gyrA, which lie in close vicinity of the active site tyrosine (residue 122). 30 Conformational changes occurring due to mutations at T86 and P104 consequently disrupt the specific interactions critical to ligand binding.
MM-PB(GB)/SA binding free energy calculations
To evaluate the free energy differences between WT and MT, MM-PB(GB)/SA methods were employed. We estimated binding energy from 125 frames of the last 5-nsec simulation trajectories and computationally predicted free energies, and contributions of various components to binding energy for different simulation systems are summarized in Table 1. The total binding free energy estimates are approximate as they do not include contribution from configurational entropy changes.
WT, wild-type; ΔEINT, internal energy; ΔEVDW, van der Waals energy; ΔEELE, electrostatic energy; ΔEGAS/ΔE MM, ΔG gas GB; ΔGSOL-NP, ecavity PB; ΔGPB, ePB; ΔGSOLV,PB, ΔG salvation; ΔGGB, eGB; HTOT,PB, ΔG binding, PB; HTOT,GB, ΔG binding, GB; M1, mutant 1 (T86I); M2, mutant 2 (T86I/P104S); PB, Poisson–Boltzmann; GB, generalized born.
In support of experimental findings, it was observed that WT gyrA has higher affinity for ciprofloxacin and binds more favorably than any of the MT forms, as indicated in Table 1. In both the MTs, T86I and T86I/P104S, a significant drop in binding energy (ΔGbinding HTOT) compared with WT gyrA was observed using both PB and GB methods. T86I is the most commonly encountered mutation leading to fluoroquinolone resistance in majority of the C. jejuni isolates. P104S mutation, which is less frequently reported relative to T86I mutation, does not seem to alter ciprofloxacin susceptibility dramatically as evident from HTOT,PB values in Table 1. Reduced binding affinities due to the mutations influence the efficiency of ciprofloxacin binding to gyrA.
Ligand binding is majorly driven by hydrophobic interactions; however, electrostatic interactions render them specific. This binding specificity lies at the core of many crucial biological processes such as enzyme catalysis and intracellular recognition. Binding affinity is a measure of strength of substrate-specific recognition. It was revealed that the electrostatic term (ΔEELE) contributes significantly to total binding free energy, as shown in Table 1. Our results reinforce the idea that these mutations in the QRDR region are a major driving force of fluoroquinolone resistance as computationally predicted binding affinities are in accordance with experimental evidence that T86I and T86I/P104S lead to significant increase in MIC values of various quinolones.13,14,31
Per-residue decomposition of binding energy
Per-residue binding energy decomposition sheds light on the key residues that play a major role in protein–ligand interaction in a molecular system and provides a semiquantitative estimate of their contribution to complex stability. We have compared the protein–ligand interaction spectra between different molecular systems of gyrA (WT and MT models), and the contribution of individual residues classified as hotspot to both polar and nonpolar interaction terms is plotted in Fig. 3. From binding energy decomposition analysis, it was revealed that residues S30, R35, P82, H83, and Y152 contribute significantly to ciprofloxacin binding with WT gyrA. A significant decrease in free energy in MT forms was largely attributed to both polar and nonpolar contributions of hotspot residues.
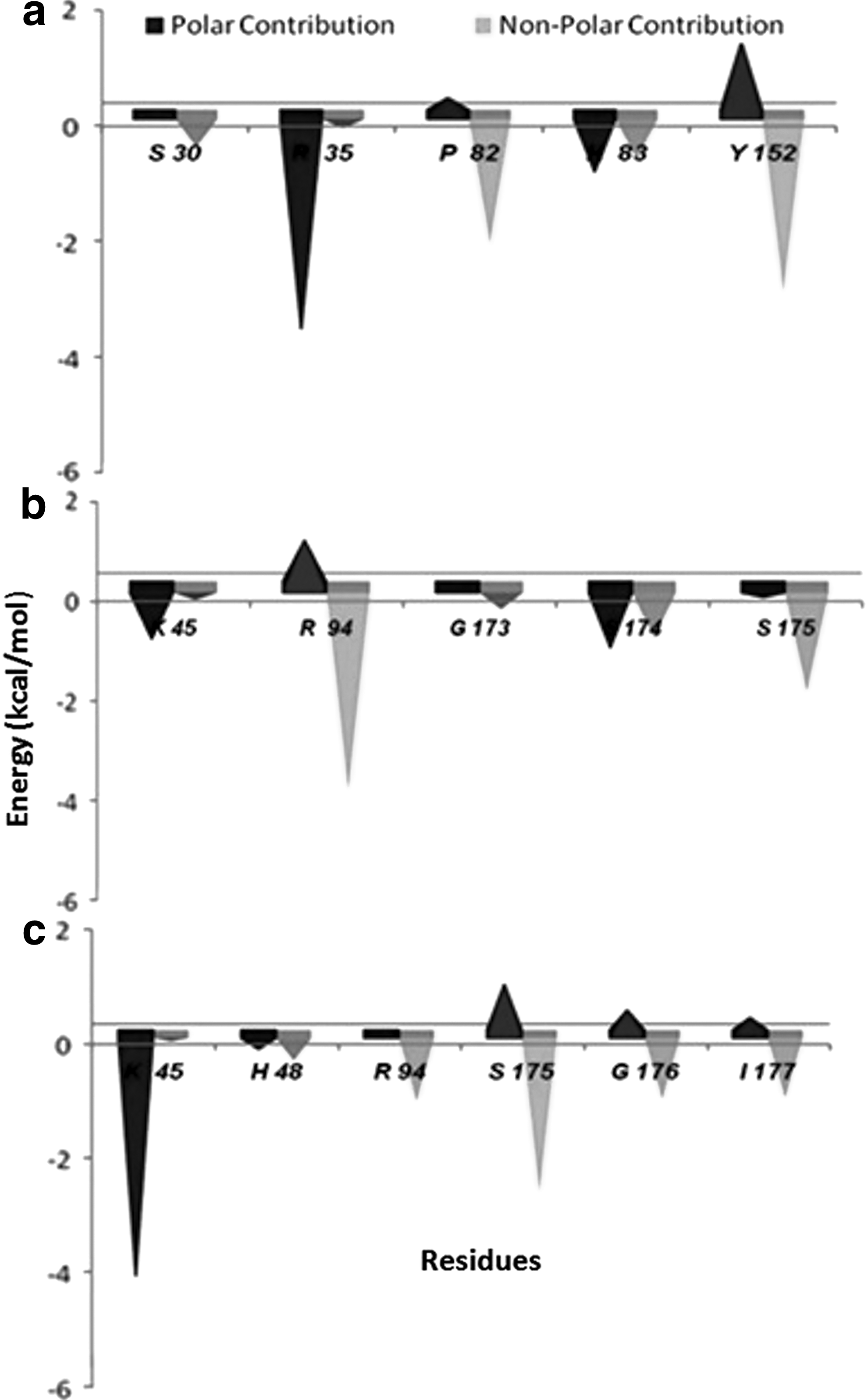
Polar and nonpolar contribution of hotspot residues from MM-Poisson–Boltzmann SA-based energy decomposition analysis of total binding energy (kcal mol−1) in
None of the hotspot residues identified in WT gyrA were reported to have significant contribution to complex stability in MTs, T86I and T86I/P104S. K45 residue in T86I/P104S has higher polar contribution compared with T86I MT, but R94 and S175 have higher contribution from van der Waals term in T86I. Although T86I/P104S showed improved binding at K45 site (total score: −4.357 kcal mol−1; electrostatic score: −33.591 kcal mol−1) likely due to its hydrogen-bonded interactions with the R49 side chain, it has a reduced total free energy compared with T86I MT (Table 1) as T86I and P104S substitution negatively impacts the binding affinity of nearby residues.
Principal component analysis
To probe the dominant motions of ensemble of conformations in WT and MT complexes, PCA was carried out on the last 5-nsec trajectories of each molecular system. We analyzed the trajectories of WT and MT gyrA in complex with ciprofloxacin to reveal the dominant motions in the conformational ensembles of WT and MT molecular systems and probe the dynamical features in each ensemble. The complexity of time evolution data of atomic coordinates was simplified by reducing it to a new coordinate system of covariance matrix to extract dominant motions. ED allows separating dominant, concerted conformational rearrangements from random conformational fluctuations.
The trajectories of ciprofloxacin-bound WT and MT complexes analyzed using PCA to identify dominant motions along the direction of first PC are plotted in Fig. 4 in the form of a porcupine plot. The porcupine plot represents major fluctuations, and significant differences in the overall pattern of global motions in WT and MT molecular systems are clearly distinguishable. In the WT, major dominant motions, which are represented by high-magnitude arrows, were mostly observed in the lower and uppermost domains that are similar to the tower domain in E. coli. These motions were of reduced magnitude and varying directions in both MTs. In T86I/P104S MT, corresponding principal motions were observed to be of the least magnitude.
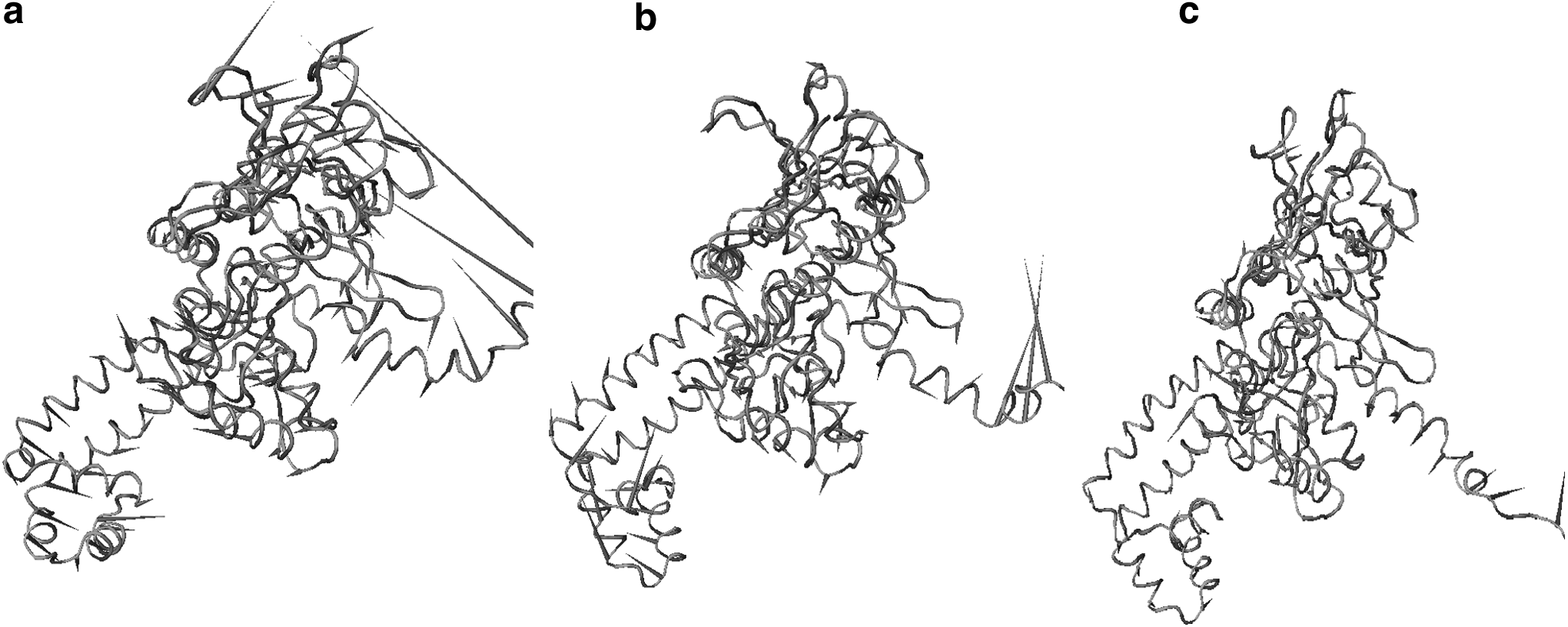
Porcupine plot of the first eigenvector obtained by principal component analysis from simulation ensembles of
Motions in the lower domain, which connects two α-helices, were also distinct among WT and MT forms of gyrA; T86I/P104S was characterized by prominently reduced motions that reflect reduced flexibility. gyrA is predicted to switch between its open and closed conformations due to the flexion of two connecting α-helices that connect the dimerization domain to C-terminal domain and tower domain. Consequently, any changes in flexibility of this domain would ultimately affect ligand binding efficiency. By and large, prominent dominant motions vary both in direction and magnitude among WT, T86I, and T86I/P104S MTs. It is apparent that WT is the most flexible of all three, which is in accord with experimental findings that report increased resistance in MT forms.
The projections along the PC reveal differences in the pattern of fluctuations for all three domains. The most distinctive motions were observed in the tower domain that harbors the QRDR responsible for quinolone resistance. Similar revelations have been made for cytochrome P450 2C9 (CYP2C9) that is crucial for excretion of over 100 prescribed drugs. However, mutations in CYP2C9 cause changes in metabolic activity, which result in adverse drug effects. 32 CYP2C9, a protein in its native state, exists as equilibrium of various conformations on an energy landscape, and studying dominant motions allows revealing the fluctuations in structural properties between different conformations induced upon ligand binding.
RIN analysis
From both structural and functional perspectives, proteins are complex molecules and their small-world behavior can be extensively studied by representing inter-residue protein contacts using graph theory in the form of RINs. RINs can capture residue interactions and find potential application in many scenarios such as identification of key residues involved in protein folding and functionally different states of protein and recently in study of protein–ligand interactions.33–35 To identify key residue interactions and explore the differences between different WT and MT models, RINs were generated using representative structures from the last 5-nsec trajectories of each molecular system and plotted in Figs. 5–7. It is apparent that RIN plots of MT forms of gyrA vary considerably when compared with the WT and from each other as well.
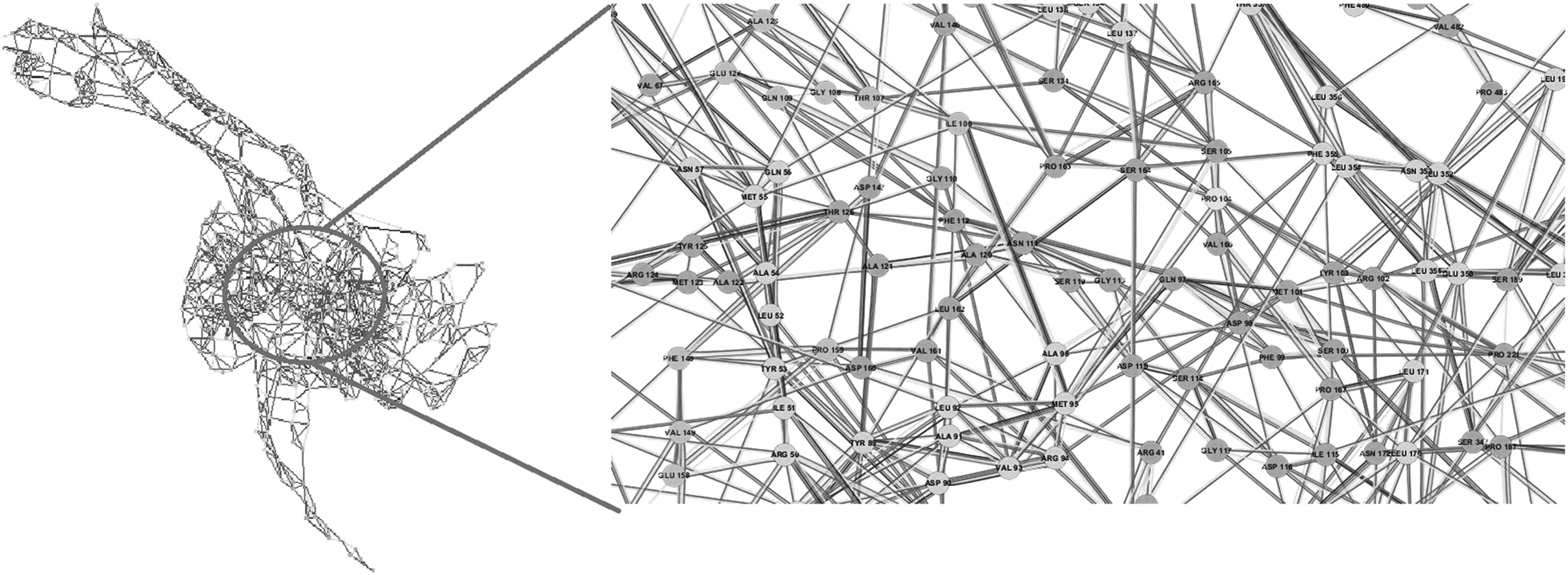
RIN map of WT. Inter-residue interactions simplify complex behavior of proteins using graph theory and facilitate the study of critical biological processes such as protein folding and protein–ligand interactions. RIN, residue interaction network. For ease of readability, figure labels can be viewed online at www.liebertpub.com/mdr
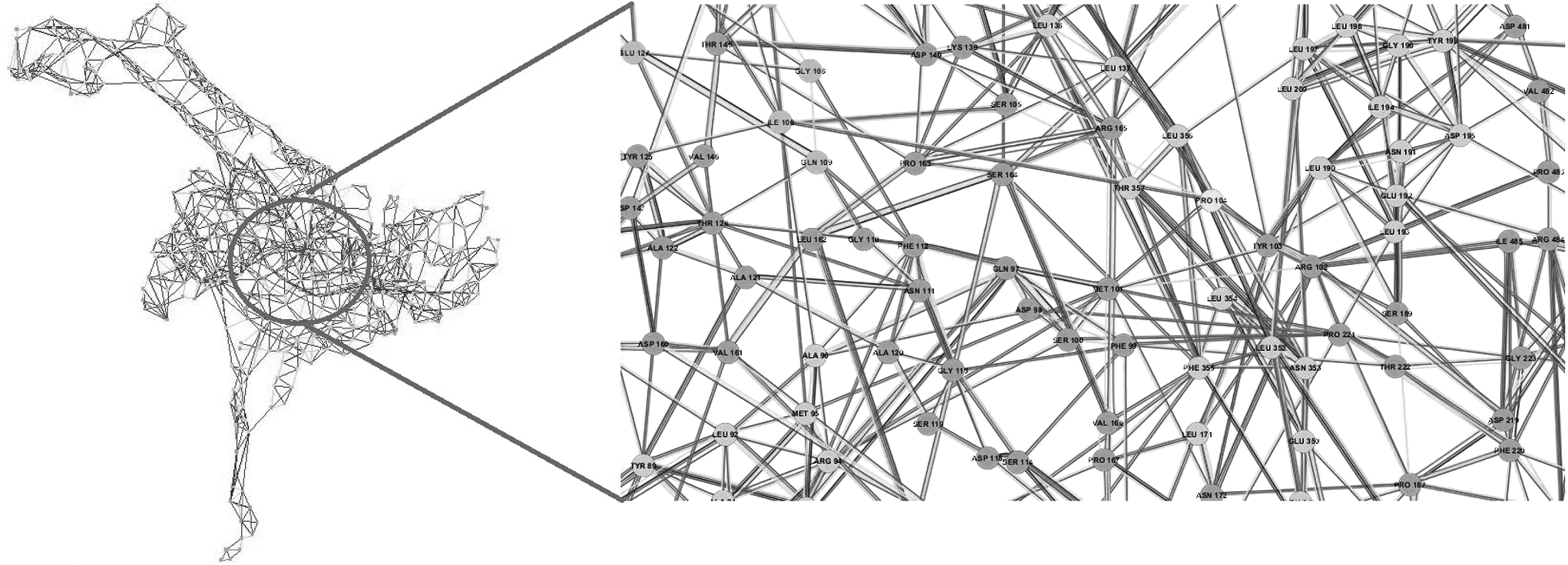
RIN map of T86I MT. Zoomed view highlights changes in the inter-residue network in the vicinity of mutated residue position 86. A single point mutation can have a huge impact on residue interaction networks. For ease of readability, figure labels can be viewed online at www.liebertpub.com/mdr
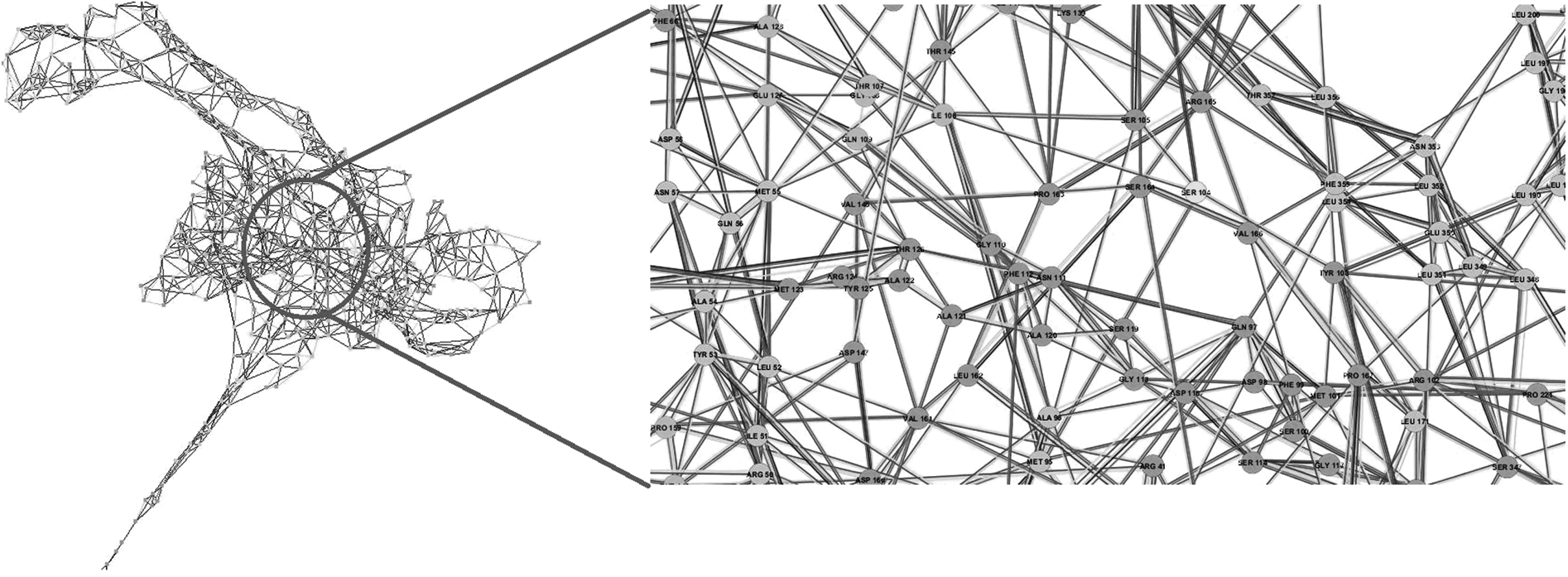
RIN map of T86I/P104S MT docked with ciprofloxacin. Zoomed view highlights changes in the network in the vicinity of mutated residue positions 86 and 104. Such mutations can lead to drastic changes disrupting active site landscape, which defines the strength of ligand binding. For ease of readability, figure labels can be viewed online at www.liebertpub.com/mdr
A zoomed-in view of the RINs shows that the interaction network in the neighborhood of mutated residue positions 86 and 104 is conspicuously discernible across WT and MT variants of gyrA. Compared with WT gyrA, as an instance, P104 in T86I MT has lost interactions with M95, D98, and S105, while S104 interactions in T86I/P104S MT with D98, M101, and R102 are lost. Similar approach has been applied to study the molecular impact of M184V mutation on HIV-1 reverse transcriptase resistance to 3TC (lamivudine). 36 This mutation is associated with a complete loss of ligand fitness and was observed to disturb the inter-RIN and ligand orientation in the active site.
Studying conserved residues and their interatomic interactions facilitates a deeper understanding of protein structure–function relationships and the critical role that the residues play in maintaining active site framework. Effect of small changes at individual residue sites adds up to extreme effect on protein structure, which will eventually lead to prominent distortion of active site moieties. It is quite interesting to observe that point mutations can significantly disrupt the RIN of a protein, subsequently affecting the drug binding landscape.
Conclusion
Significant advancements in computer processing competency have rendered investigations in the effect of mutations at the atomistic level possible. We performed a comparative MD simulation study of WT, T86I, and T86I/P104S MT complexes of gyrA and ciprofloxacin in C. jejuni to gain insights into the atomistic details of underlying quinolone resistance in MT form of gyrA. The absence of large fluctuations in RMS deviation, RMS fluctuation, and secondary structure conservation points toward the idea that WT and MT trajectories were exploring the same configurational space and that no significant changes in backbone conformation were observed.
Despite that, incorporated mutations significantly impact the dynamic behavior of complexes, which was reflected by major disruptions in the RIN and altered dominant motions that could destabilize active site conformation reducing ligand binding efficiency. This study offers reassuring consistency with experimental observations of increased MIC values due to these mutations and provides a comprehensive picture of genotype–phenotype association.
Acknowledgments
This research received funding support from the FAST TRACK Young Scientist Fellowship from the Department of Science and Technology (DST), Ministry of Science and Technology, India, under grant number SB/FT/LS-278/2012. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.