
Abstract
Antimicrobial resistance is common in the microbial inhabitants of the human oral cavity. Antimicrobials are commonly encountered by oral microbes as they are present in our diet, both naturally and anthropogenically, and also used in oral healthcare products and amalgam fillings. We aimed to determine the presence of genes in the oral microbiome conferring reduced susceptibility to common antimicrobials. From an Escherichia coli library, 12,277 clones were screened and ten clones with reduced susceptibility to triclosan were identified. The genes responsible for this phenotype were identified as fabI, originating from a variety of different bacteria. The gene fabI encodes an enoyl-acyl carrier protein reductase (ENR), which is essential for fatty acid synthesis in bacteria. Triclosan binds to ENR, preventing fatty acid synthesis. By introducing the inserts containing fabI, ENR is likely overexpressed in E. coli, reducing the inhibitory effect of triclosan. Another clone was found to have reduced susceptibility to cetyltrimethylammonium bromide and cetylpyridinium chloride. This phenotype was conferred by a UDP-glucose 4-epimerase gene, galE, homologous to one from Veillonella parvula. The product of galE is involved in lipopolysaccharide production. Analysis of the E. coli host cell surface showed that the charge was more positive in the presence of galE, which likely reduces the binding of these positively charged antiseptics to the bacteria. This is the first time galE has been shown to confer resistance against quaternary ammonium compounds and represents a novel, epimerase-based, global cell adaptation, which confers resistance to cationic antimicrobials.
Introduction
A
It is estimated that more than half of oral bacteria cannot be grown in a laboratory environment. 2 Therefore, to analyze the entire metagenome for resistance, we can clone it into a replicon in a heterologous bacterial host such as E. coli and screen for gain of phenotype. Functional metagenomics is a powerful technique for the identification of antibiotic resistance genes, which have been performed on various environmental samples such as soil, water, and the human microflora.3–5 Novel antibiotic resistance genes have also been isolated by using functional metagenomics, for example, the chloramphenicol efflux gene pexA from Alaskan soil, a novel gentamicin resistance gene from river sediment samples, 10 novel β-lactamase gene families from the human gut microbiome, the tetracycline resistance gene tet(37), and the tetracycline and tigecycline resistance gene tetAB(60) from the human oral cavity.4–8
In this work, we aimed to detect antimicrobial resistance genes from the human oral metagenome. We chose a selection of antimicrobials, with different mechanisms of action, which oral bacteria are likely to come into contact with regularly. These include metals present in amalgam fillings, antiseptics widely used in oral healthcare products, and preservatives used in food production.
We have identified individual clones exhibiting reduced susceptibility to triclosan, cetylpyridinium chloride (CPC), and cetyltrimethylammonium bromide (CTAB) and determined the likely mechanisms responsible.
Materials and Methods
Construction of human oral metagenomics library
Human oral metagenomics DNA was extracted from the saliva samples collected from 11 healthy individuals, both males and females, from the Department of Microbial Diseases, University College London (UCL) Eastman Dental Institute. None of the volunteers had received antibiotics for at least three months before the saliva collection date. This project received ethical approval from UCL Ethics Committee (project number 5017/001), and written consent forms were collected from the volunteers. The oral metagenome was extracted as described previously. 9 The library was then constructed with pCC1BAC vector and transformed into TransforMax EPI300 Electrocompetent E. coli and individual clones stored in microtiter plates at −80°C, as described previously. 7
Library screening and minimum inhibitory concentration determination
The minimum inhibitory concentration (MIC) of antimicrobial compounds was determined against E. coli EPI300 containing pCC1BAC vector to be used as the concentrations for the screening. The MIC determination was performed by following the broth microdilution method described previously. 10 The bacterial strains were cultured in Luria-Bertani (LB) broth containing chloramphenicol (12.5 μg/ml) and incubated overnight in 37°C shaker. The overnight culture was diluted to OD600 of 0.1. In a 96-well plate, 90 μl of LB broth, containing 12.5 μg/ml chloramphenicol and different concentration of antimicrobials (listed in Table 1), and 10 μl of diluted E. coli were added to the wells to make a total 100 μl volume. The plates were then incubated on a shaker at 37°C for 18 hr and checked for their growth to determine the MIC breakpoints.
CTAB, cetyltrimethylammonium bromide; CPC, cetylpyridinium chloride; MIC, minimum inhibitory concentration.
Each 96-well plate of the human oral metagenomic DNA library E. coli was then screened against various antimicrobial substances with respect to the determined MIC. An ethanol sterilized 96-pin replicator was used to inoculate the E. coli library into a 96-well plate containing 100 μl of LB broth containing chloramphenicol and antimicrobials. The inoculated plates were then incubated in a shaking incubator at 37°C overnight.
Plasmid extraction and sequencing
Clones of interest were subcloned into 5 ml of LB broth containing chloramphenicol and the antimicrobials that they showed resistance to, and incubated in a 37°C shaker for 18 hr. The copy number induction culture was then set up to induce the number of the pCC1BAC plasmid in each cell from a single copy up to 25 copies per cells by transferring 1 ml of overnight culture into 9 ml of LB broth supplement with chloramphenicol (12.5 μg/ml) and CopyControl™ Induction Solution (10 μl in 10 ml), and then incubated in a 37°C shaker for 5 hr. Plasmids were extracted by the QIAprep Spin Miniprep kit (Qiagen, United Kingdom) following the manufacturer's instructions. The size of inserts was estimated by digesting the plasmids with HindIII restriction enzyme (New England Biolabs, United Kingdom), following the manufacturer's instructions, and the DNA fragments were visualized on 1% agarose gel stained with ethidium bromide. The inserts on the extracted plasmids were initially sequenced from both ends with pCC-F and pCC-R primers by using the service from Genewiz (Genewiz, United Kingdom). Additional primers were designed and used for the subsequent sequencing to extend the sequences of the inserts (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/mdr).
Transposons mutagenesis
To locate the genes conferring resistance, transposon mutagenesis was performed by using the Template Generation System II Kit (Thermo Scientific, United Kingdom), following the manufacturer's instructions. Entranceposon (KanR-3) was selected for the mutagenesis. The reaction mixture was electroporated into E. coli EPI300 (Cambio, United Kingdom) and grown on LB agar with chloramphenicol (12.5 μg/ml) and kanamycin (20 μg/ml). Each colony was then subcultured into 96-wells plates containing LB broth containing chloramphenicol (12.5 μg/ml), kanamycin (20 μg/ml), and the corresponding resistant antimicrobials (8 μg/ml CTAB for A10F2 and 0.06 μg/ml triclosan for C7C4). After incubating overnight in a 37°C shaker, clones that lost the resistant phenotype were selected for the extraction of their plasmids and sent for sequencing with SeqE and SeqW primers, provided with the kit to determine the target sites.
Detection of fabI on the DNA inserts of triclosan-resistant clones
The presence of the fabI gene on the DNA inserts of each triclosan-resistant clone was carried out by using a rational polymerase chain reaction (PCR) approach. Primers were designed to target fabI gene from different bacterial species, identified by the end sequencing of each clone (Supplementary Table 1). The PCR reaction contained 15 μl of 2 × BioMix Red (Bioline, United Kingdom), 0.2 μM of each primer, 50–100 ng of extracted plasmid from triclosan-resistant clones, and molecular grade water (Sigma, United Kingdom) up to 30 μl. The PCR cycle was programmed as follows: initial denaturation at 94°C for 3 min, 35 cycles of (i) denaturation at 94°C for 1 min, (ii) annealing at 54–58°C for 30 sec, and (iii) extension at 72°C for 1 min, and final extension at 72°C for 3 min. The PCR products were then purified using the QIAquick PCR Purification Kit (Qiagen, United Kingdom), following the manufacturer's protocols, and confirmed by sequencing.
Subcloning of UDP-glucose 4-epimerase and glucose-6-phosphate isomerase genes
The primers, listed in Supplementary Table 1, were designed to amplify the UDP-glucose 4-epimerase and glucose-6-phosphate isomerase (GPI) genes, identified from the transposon mutagenesis. The PCR reaction contained 15 μl of 2 × BioMix Red (Bioline, United Kingdom), 0.2 μM of each primer, 50–100 ng of A10F2 plasmid, and molecular grade water (Sigma, United Kingdom) up to 30 μl. The PCR cycle was programmed as follows: initial denaturation at 94°C for 3 min, 35 cycles of (i) denaturation at 94°C for 1 min, (ii) annealing at 54–58°C for 30 sec, and (iii) extension at 72°C for 1 min, and final extension at 72°C for 3 min. The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, United Kingdom), following the manufacturer's protocols. The amplicons were digested with EcoRI and HindIII restriction enzymes (New England Biolabs, United Kingdom) and directionally cloned into predigested pCC1BAC vector. The ligation mixture was electroporated into electrocompetent E. coli EPI300 and grew on LB agar supplemented with chloramphenicol (12.5 μg/ml), 0.4 mM IPTG, and 40 μg/ml Xgal. Clones with inserts were selected and confirmed by extracting the plasmids and sequencing across the cloning site.
Cytochrome c binding assay
The bacterial surface charge was determined by performing a cytochrome c binding assay as described previously with some modifications.
11
Bacterial cells were collected at the mid-log phase and washed twice with 20 mM MOPS buffer (Sigma, United Kingdom) at pH 7. The cells were then serially diluted to the final OD600 ranging from 1 to 7 and resuspended in 150 μg/mL cytochrome c (Sigma, United Kingdom). After that, the cells were incubated at room temperature with shaking at 200 rpm for 10 mins and centrifuged at 5,000 rpm for 10 mins. The absorbance of the supernatant was then measured at 530 nm. The amount of cytochrome c bound to the cells was calculated by comparing the absorbance values to the 150 μg/ml cytochrome c stock solution. The amount of unbound cytochrome c was calculated by
The average and standard deviation of the amount of cytochrome c bound to the cells were calculated from three biological replicates, which were used for the columns and error bars, respectively. Statistical analysis was carried out by using t-test to determine whether the amounts of cytochrome c bound to E. coli::pCC1BAC and E. coli::[pCC1BAC::galE] at each cell density were statistically significantly different with p-value <0.05 (*) or p-value <0.005 (**).
Results
Functional screening for antimicrobial resistance genes
Before the screening, the MIC breakpoints for various antimicrobials against E. coli EPI300 containing pCC1BAC vector were determined by broth microdilution method. The results are shown in Table 1, which were used as the concentrations for the library screening. Out of 12,227 clones screened, 10 clones were confirmed for their reduce susceptibility to triclosan, (clones A5A12, A5B12, C7C4, E4E3, H4E9, L5F10, M8G5, N9E7, R2G11, and X5F8) and another one to CTAB (clone A10F2).
Characterization of genes conferring reduced susceptibility to triclosan
The identification first focused on the C7C4 clone. End sequencing of C7C4 plasmid showed that it had homology to Neisseria meningitidis (accession number CP012392.1). The size of C7C4 DNA insert was determined by digesting plasmid with HindIII restriction enzymes and visualized on 1% agarose gel (Supplementary Fig. S1A). Four DNA fragments were shown with the total estimated size of 24.7 kb. As the C7C4 insert DNA was large, the gene conferring triclosan resistance was then identified by transposon mutagenesis. Six clones with the loss of triclosan-resistant phenotype were found. Sequencing results of mutant clones showed that transposons inserted into fabI gene on C7C4 insert (Fig. 1).
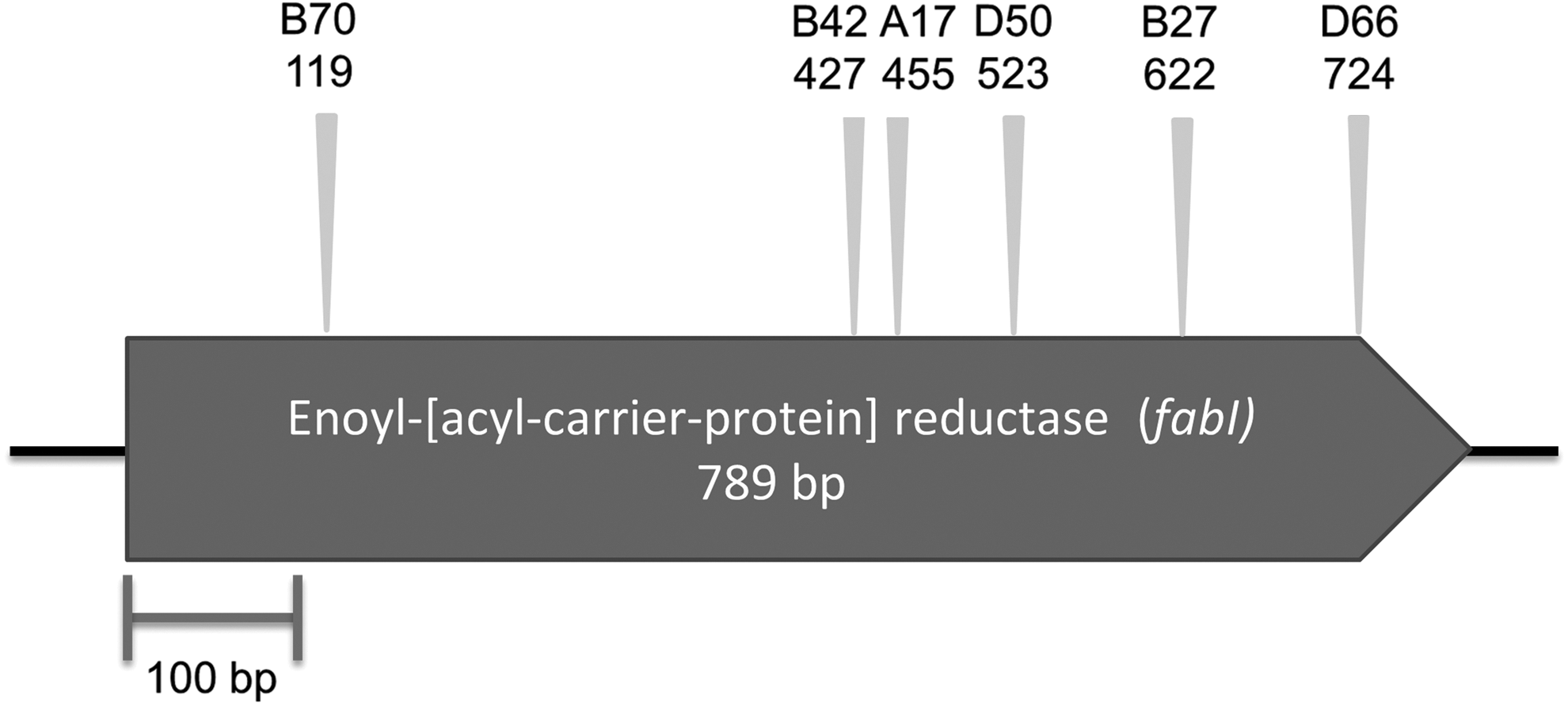
The putative gene responsible for reduced triclosan susceptibility in C7C4 clone identified by transposon mutagenesis. The name of mutant clones and the position of the transposons are indicated with triangles. The open arrowed boxes represent ORFs, pointing in the probable direction of transcription.
Primers specific for N. meningitidis fabI were designed and used for the PCR on the plasmid templates of the other triclosan-susceptible clones to determine whether they carried the same gene as C7C4. No product was found from these PCRs. These plasmids were end sequenced revealing the likely source of DNA, which were Campylobacter concisus (clones A5A12 and M8G15: accession number CP000792.2), Campylobacter gracilis (clone A5B12: accession number CP012196.1), Prevotella sp. (clones E4E3, L5F10 and X5F8: accession number CP002122.1), Haemophilus parainfluenzae (clones N9E7 and R2G11: accession number FQ312002.1), and Porphyromonas sp. (clone H4E9: accession number WP_044114258.1).
By mapping the end sequencing to the genomes, we were able to show that the inserts likely contained the cognate fabI. This was confirmed by carrying out PCR using primers targeting these individual fabI followed by sequencing. The sequence analysis of these partial fabI amplicons (229–376 bp) also showed that none of the fabI was 100% identical to the fabI in the genomes from where the primers were designed. The percentage identity ranged from 86% to 98%. The gene fabI encodes enoyl-acyl carrier protein reductase (ENR), which is an enzyme involved in type II fatty acid synthesis. As triclosan blocks the fatty acid synthesis by binding to ENR, the reduced susceptibility of these clones is likely to be a result of either the overexpression of ENR from the extra copy of fabI within the plasmids or the expression of a fabI encoding ENR with lower susceptibility to triclosan.
Characterization of genes conferring reduced susceptibility to quaternary ammonium compounds
The plasmid from the A10F2 clone was extracted and digested with HindIII to determine the size of the insert, which was estimated to be 17.1 kb (Supplementary Fig. S1B). The extracted plasmid was also sent for sequencing across the cloning site. The sequencing results revealed that the insert was likely to be chimeric with respect to the source of the DNA. The region from one end was most closely related to Veillonella parvula (accession number CP001820.1, 85% nucleotide identity) and the sequences on the other end were mostly related to Prevotella melaninogenica (accession number CP002122.1, 80% nucleotide identity).
Transposon mutagenesis was performed on the plasmid to determine the gene(s) responsible for the reduced susceptibility to CTAB. Ten clones with gain of sensitivity to CTAB were selected and plasmids were sent for sequencing using primers located at the end of transposons. The sequencing results showed that nine inserts were located in a UDP-glucose 4-epimerase (GalE) gene, and one was inserted in a gpi gene, immediately upstream of galE (Fig. 2A). This region of the insert, encompassing the isomerase and the epimerase, showed 87% nucleotide identity to Veillonella parvula (accession numbers; CP019721.1 and CP001820.1). The nucleotide sequence was deposited in the nucleotide database with the accession number KY769203.
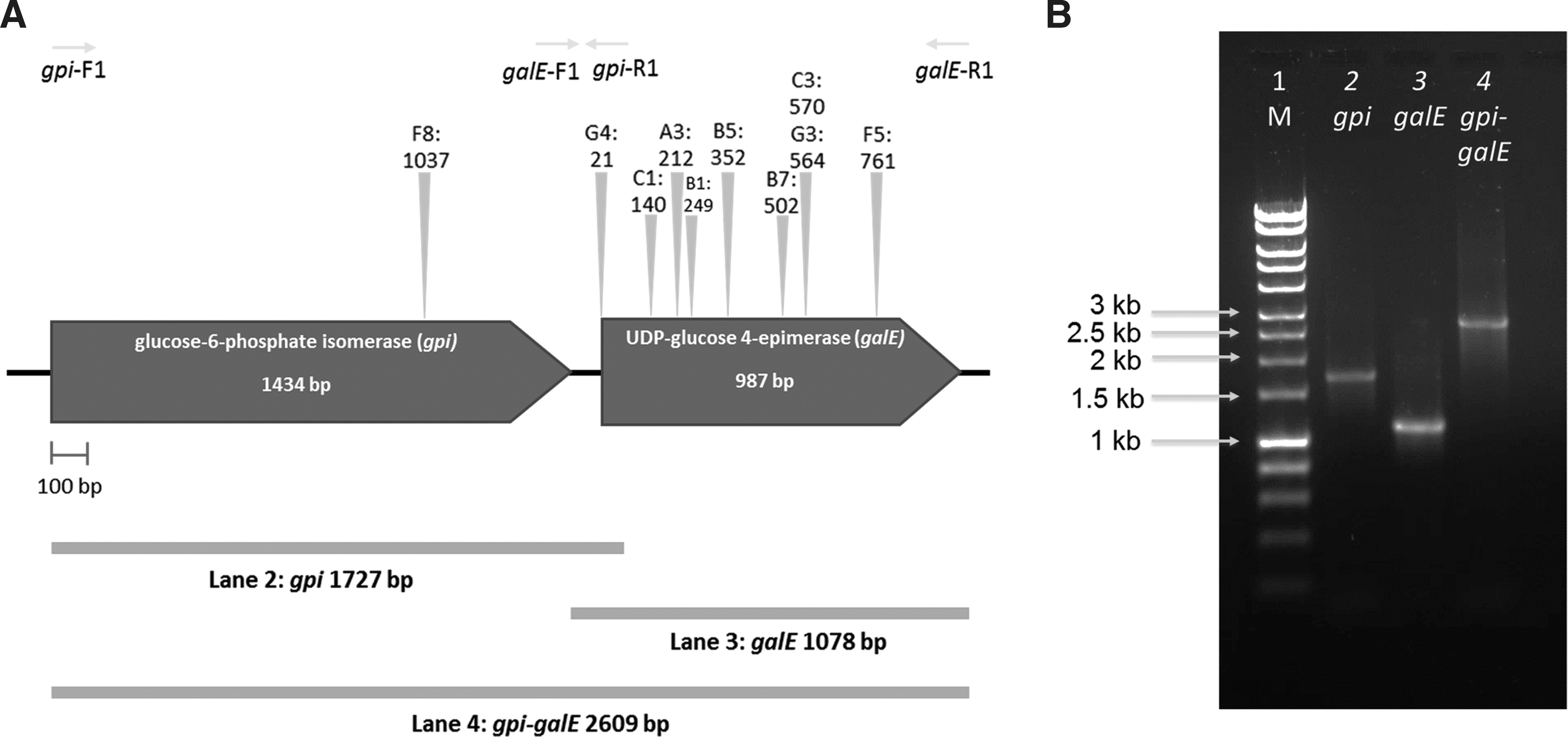
To confirm the function of the putative genes conferring reduced susceptibility to CTAB, galE, gpi, and gpi-galE were amplified from the A10F2 plasmid (Fig. 2B), ligated into pCC1BAC vector and electroporated into E. coli EPI300. Only pCC1BAC::galE and pCC1BAC::gpi-galE E. coli showed reduced susceptibility to CTAB, suggesting that only galE was responsible for the CTAB resistance in E. coli. The MIC value of CTAB against pCC1BAC::A10F2, pCC1BAC::gale, and pCC1BAC::galE-gpi E. coli increased twofold from 8 to 16 μg/ml (Fig. 3). As the mode of action of CTAB is similar to CPC, the MIC of CPC were then determined and shown to increase from 6 to 8 μg/ml. 12 This is the first report of galE being responsible for reduced susceptibility to quaternary ammonium compounds (QACs).
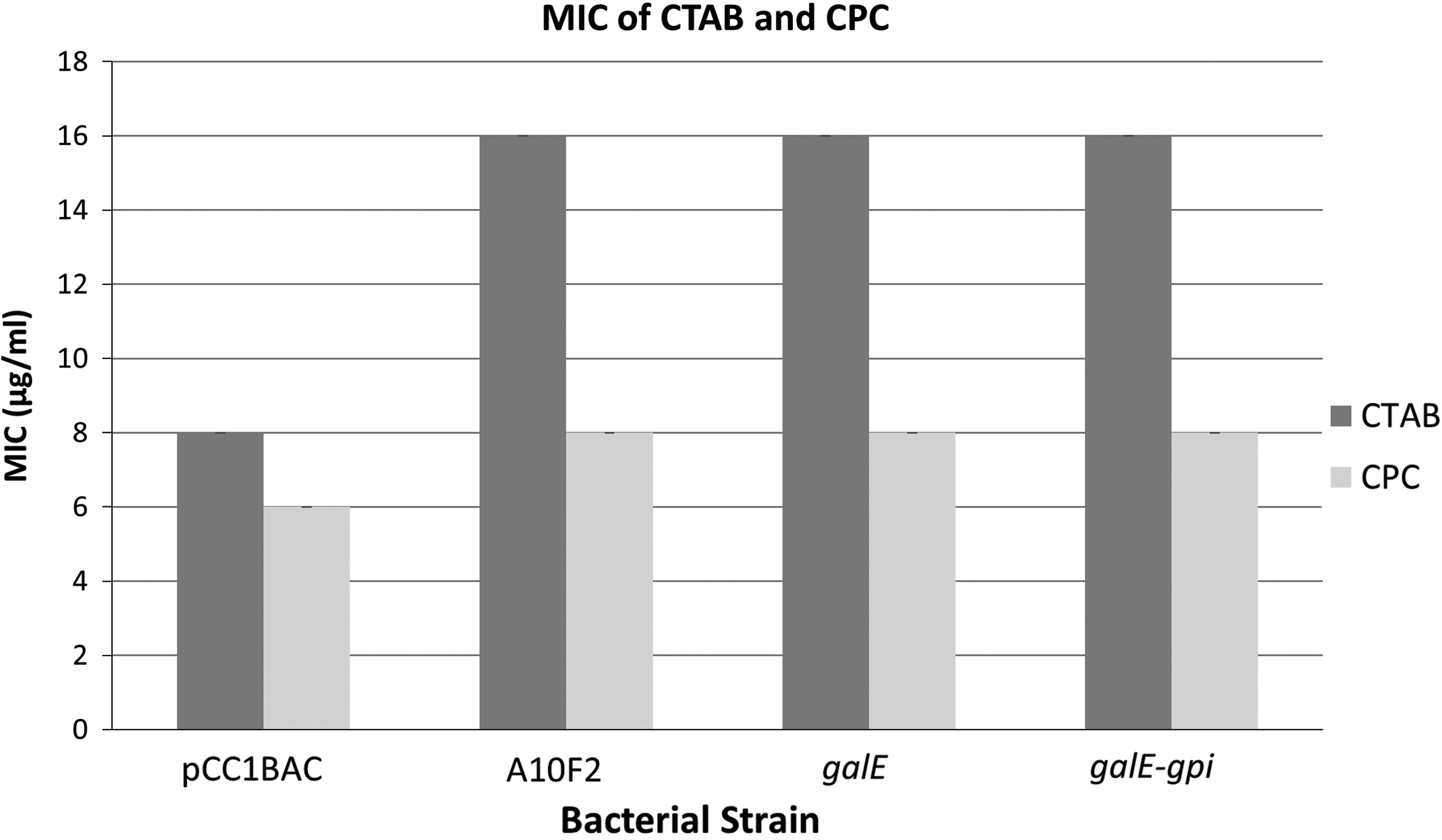
Minimum inhibitory concentration of CTAB and CPC against Escherichia coli::pCC1BAC, E. coli::[pCC1BAC::A10F2], E. coli::[pCC1BAC::galE], and E. coli::[pCC1BAC::galE-gpi] determined from three replicates of broth microdilution. CPC, cetylpyridinium chloride.
Comparison of the amino acid sequences of GalE from A10F2 with the databases showed homology to GalE from V. parvula (accession number WP_060918982.1) and E. coli (accession number EFJ53418.1) with 97% and 56%. An alignment of the protein sequences showed that they all had the nicotinamide adenine dinucleotide (NAD) binding sites and substrate binding sites, which are essential for the epimerase activity, at a similar position (Fig. 4).
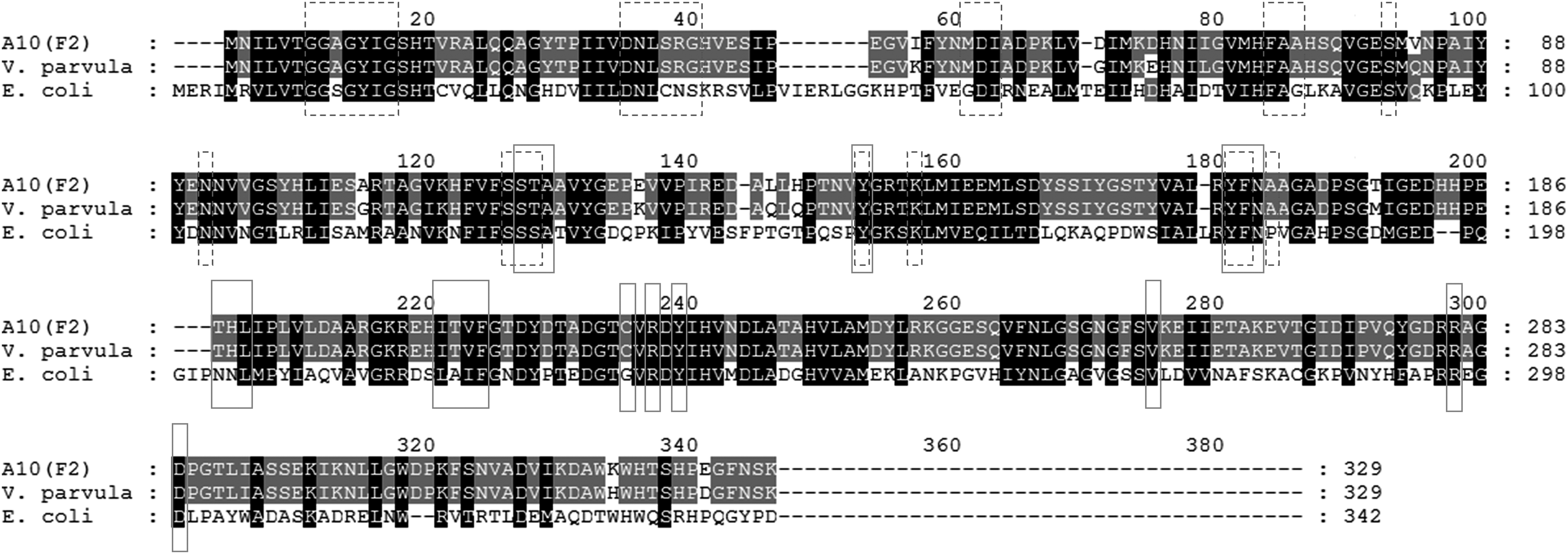
The alignment of galE amino acid sequences from the A10F2 clone, V. parvula, and E. coli. The dashed and solid boxes indicate the NAD binding site and substrate binding site, respectively. The black and gray shading indicate the base pairs with 100% and 80% conservation level. NAD, nicotinamide adenine dinucleotide.
Bacterial cell surface charge determination
GalE catalyzes the reversible epimerization of UDP-glucose to UDP-galactose and the interconversion of UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine,13,14 which are necessary for the biosynthesis of lipopolysaccharide (LPS). LPS is responsible for the overall negative charge of a gram-negative cell outer membrane. The cell surface charge of E. coli::pCC1BAC and E.coli::[pCC1BAC::galE] was therefore determined by using a cytochrome c assay. We demonstrated that lower amount of cytochrome c bound to the E. coli::[pCC1BAC::galE], compared to E. coli::pCC1BAC (Fig. 5). As cytochrome c is a cationic peptide that can bind to the negatively charged bacterial cell surface, the reduced binding of cytochrome c to E. coli::[pCC1BAC::galE] indicates that the cell surface is more positively charged in this clone. As CTAB and CPC are both positively charged compounds, we can infer that they would bind less to the E. coli::[pCC1BAC::galE] cell surface.

The bar chart represents the amount of cytochrome c bound to pCC1BAC and galE E. coli at the cell density from OD600 of 1–7. The error bars represent standard deviation from three biological replicates. The asterisks indicate the statistically significant difference between the amount of cytochrome c bound to E. coli::pCC1BAC and E. coli::[pCC1BAC::galE] at each cell density with the p-value <0.05 (*) and <0.005 (**) determined by using t-test.
Discussion
Triclosan is one of the most common ingredients used in personal hygiene products, such as soaps, deodorants, and toothpastes. Its antiseptic activity is the inhibition of fatty acid synthesis by targeting ENR. The binding of triclosan to ENR dramatically increases the binding affinity of ENR to NAD+, resulting in the formation of an ENR-NAD+-triclosan ternary complex.15,16 This formation blocks ENR from participating in the synthesis of fatty acids, which is required for building the cell membrane. The compound was recently banned by the Food and Drug Administration from consumer antiseptic wash products. 17
Previous mechanisms of triclosan resistance include mutations in fabI, inhibiting the formation of the ENR-NAD+-triclosan ternary complex in Staphylococcus aureus, E. coli, and Mycobacterium tuberculosis.18–20 Overexpression of fabI is another mechanism of triclosan resistance and can be as a result of an increased expression of a native gene, or acquisition of an additional copy on a mobile genetic element.21,22 In one study, the acquisition of fabI in S. aureus was shown to be facilitated by IS1272 composite transposons, designated TnSha1 and TnSha2, from Staphylococcus haemolyticus. 22
QACs, including CTAB and CPC, are cationic surfactants, containing a centrally charged nitrogen with four alkyl or aryl groups. 23 One of the important characteristics of QACs is their broad antimicrobial activity against bacteria, yeasts, fungi, and viruses. Coupled with their low toxicity, QACs are used widely as disinfectants in various industries and environments, such as food processing, cleaning products, and healthcare environments. 24 Their antimicrobial activity begins with the absorption to, and penetration into the cell wall, followed by disorganization of the cell membrane leading to leakage and degradation of intracellular material, and subsequent cell lysis. 12
Several studies on QAC resistance have been reported previously, including resistance in Listeria and Salmonella detected in food industries.25–27 In most cases, an increase in MIC was found, but this was still below the concentration used in industries or no cross-tolerance to other antimicrobials has been detected. The transfer of a CTAB resistance gene (qrg) on a Tn916-like conjugative transposon between streptococci, by transformation, has also been demonstrated. 28
UDP-glucose 4-epimerase (GalE) is a ubiquitous enzyme, catalyzing the interconversion of UDP-glucose and UDP-galactose in the Leloir pathway of galactose metabolism, and the interconversion of UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine in glycoprotein and glycolipid synthesis.13,14 UDP-galactose, UDP-glucose, and UDP-N-acetylglucosamine are also substrates in the LPS biosynthesis pathway, which depend on the activity of GalE. 29 LPS is a negatively charged molecule, and is a major component of the outer membrane of gram-negative bacteria. Therefore, changing the structure and quantity of LPS on the membrane can result in the alteration of the bacterial surface charge. Previously, a mutation of galE in Campylobacter jejuni was shown to reduce the molecular weight of LPS lipid A-core and also the ability of the bacteria to adhere and invade human INT 407 (HeLa derivative) cells. 30
Our results showed that the cell surface charge was less negative due to the presence of the cloned galE, possibly from V. parvula. An alignment of GalE of V. parvula and E. coli suggested that they were similar and would be able to function in E. coli. Expressing a heterologous galE gene, possibly from V. parvula in E. coli, will likely result in a higher epimerase activity in the cells. As the reactions catalyzed by GalE are reversible, the overexpression of a heterologous epimerase is likely to affect the balance of substrates due to altered enzyme kinetics during LPS production, which will ultimately affect cell surface charge.
The alteration of cell surface charge was previously shown to confer resistance to daptomycin, a cationic antimicrobial peptide. Mutations in genes involved in cell wall homeostasis and cell membrane phospholipid metabolism in Enterococcus faecalis and S. aureus affected the cell surface, resulting in it becoming more positively charged, which repelled daptomycin, reducing binding to the cell membrane.31,32
Overexpression of fabI was previously shown to not only confer resistance to triclosan but also to isoniazid, an antituberculosis agent.33,34 We have shown in this study that overexpression of the epimerase GalE not only confers a reduced susceptibility to CTAB but also to CPC. An increase in tolerance to antimicrobials has been recently reported to pave the way for subsequent evolution of resistance. 35 Therefore, the acquisition of these genes may provide a window of opportunity for the development of resistance to these, and other, compounds. Multispecies biofilms, such as those that are found in the oral cavity (e.g., dental plaque), are highly conducive to horizontal gene transfer and the continued exposure of these bacteria to this myriad antimicrobial compounds provides a very strong selective pressure, should such an acquisition event occur. 36
In conclusion, we have determined that the reduced susceptibility to QACs and triclosan can be conferred by the expression of heterologous housekeeping genes in E. coli.
Footnotes
Acknowledgments
We thank Mark Webber (Institute of Food Research) for useful discussions and providing triclosan. We thank Andrew Smith (UCL Eastman Dental Institute) for useful discussions and providing sodium benzoate. L.R. was jointly funded by a UCL IMPACT studentship at UCL and by the Seedcorn Programme at the Animal and Plant Health Agency. K.R. was supported by the Erasmus programme. L.C.W. was supported by a Harry Smith Vacation Studentship from the Microbiology Society.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.