
Abstract
The family Legionellaceae consists of Gram-negative bacteria that are widely distributed in aquatic environments around the world. This family consists of a single genus, Legionella, that is recognized as an important cause of community-acquired pneumonia and hospital-acquired pneumonia. Legionella consists of intracellular pathogens, thus cellular pharmacokinetic and pharmacodynamic properties of an antibiotic against these bacteria as well as uptake and subcellular distribution into macrophages should be considered for a successful outcome of disease. Treatment strategies for Legionella infection require a combination of multiple antibiotics. Hence, because of the possible development of resistance to the drugs during therapy, a new alternative targeted therapy is yielding promising results. In this study, a comprehensive in silico target identification pipeline was performed on members of the family Legionellaceae to identify the best targets. Using a homology-based computational pipeline method, new drug targets were identified. Of 4,358 analyzed proteins, 18 proteins, including proteins involved in metabolism (amino acid, energy, and lipid metabolisms), cellular transport, cell division, and cell motility, were selected as the final putative drug targets. These proteins play an important role in the survival and propagation of Legionella infection. In conclusion, homology-based methods could improve the identification of novel drug targets and the drug discovery process, which can potentially be effective for the prevention and treatment of Legionella infections.
Introduction
T
Pneumonia due to Legionella infection is an important public health problem that is frequently caused by Legionella pneumophila with either community- or hospital-acquired Legionellosis. Therefore, this organism has the potential to cause both outbreaks and sporadic cases.5,6 The fatality rates associated with Legionnaires' disease among untreated patients are ∼15–20%. 7 Owing to the small size of Legionella and adaptation to intracellular compartments, an appropriate treatment of the antimicrobial agents is crucial for a successful outcome of Legionnaires' disease; on this basis, intracellular penetration, accumulation, and distribution are important parameters to the activity of antibiotics against intracellular bacteria. 8 Thus, antimicrobial agents with adequate intracellular penetration into alveolar macrophages as well as subcellular compartments are more effective than antibiotics with poor intracellular penetration.
There are several major classes of antibiotics, including macrolides, fluoroquinolones, tetracyclines, rifamycins, and ketolides, which are used for the treatment of Legionella infections.5,7 Because of the ability of Legionella spp. to survive and multiply in human macrophages, consequently, like other intracellular pathogens, antibiotics that penetrate cell membrane are drugs of choice. Aminoglycosides in general are too water soluble and penetration into biologic membranes is poor; therefore, they are taken up slowly by endocytosis, which results in an exclusively lysosomal localization. Whereas both macrolides and fluoroquinolones accumulate in phagocytes. In addition, some drugs are quickly cleared from phagocytes, consequently their concentration and intracellular distribution vary because of differences in their intracellular pH gradients of the cytosol and subcellular compartments.7,8
On the contrary, treatment of Legionella infection requires a combination of multiple antibiotics, thus this may pose a concern on the development of drug resistance during therapy. In recent years, several cases of drug resistance to routinely prescribed antimicrobials in Legionella isolates have been reported, among which L. pneumophila strains were more resistant than other Legionella species.7,9–14 It is very important to note that hidden resistance cases have also been reported and have been suggested to be correlated with treatment failure and poor prognosis in Legionnaires patients. 15
The current strategies to identify drug targets are relatively difficult, time consuming, and expensive, often resulting in very finite drug targets.16–18 Furthermore, the stages of drug discovery and development using traditional methods are a linear multistep process and yield few drug targets with little feedback from intracellular information for guiding target selection. Therefore, with the arrival of genome research during the past decade, focus on drug development has shifted to computational comparative genomics to identify novel drug targets.16,19
In silico drug target identification, which includes a series of algorithms to identify genes and proteins, is applied to the discovery and development of potential therapeutic targets. These approaches increase the efficiency of therapeutic targets, reduce timelines and costs of the drug discovery process, and also provide a closer look of the entire microorganism. Speed, low cost, and, even more importantly, providing a systematic view of the whole microorganism are the advantages of in silico techniques for identification and validation of drug targets. These features provide a great chance for asking questions that are often difficult to address experimentally. There has been an increase in the use of in silico identification methods to find drug targets in several studies.17,18,20,21
With the help of genome-sequencing projects of pathogens and humans, microbial drug target identification has been revolutionized in recent years. Among genomic strategies, subtractive genomics has been successfully used in the identification of microbial drug targets. 22 Various methods can be used to discover a proper therapeutic target such as comparative genomics models, 23 structure and sequence to function,24,25 metabolic pathways, 26 and data mining. 27 The most used methods to identify new therapeutic targets are based on the sequence similarity using the Basic Local Alignment Search Tool (BLAST) algorithm. 28 In this study, a similarity-based method was used for genome-wide prediction of the drug targets in major pathogens of the family Legionellaceae, considering the L. pneumophila str. Paris as an important pathogen.
Materials and Methods
Step I
The complete proteome of L. pneumophila str. Paris that comprises 3,012 chromosomal proteins and 134 plasmid proteins was downloaded from the National Center for Biotechnology Information (NCBI) genome database. Virulence Factor Database (VFDB) is a practical database of virulence factors (VFs) for bacterial pathogens and also Chlamydia and Mycoplasma. All the major VFs of L. pneumophila str. Paris (127 proteins) were retrieved from VFDB. All literature reported that L. pneumophila str. Paris resistance proteins were downloaded from NCBI.21,29 Using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) 10.0 tool and seven active prediction methods (neighborhood, gene fusion, co-occurrence, coexpression, experiments, databases, and text mining), interaction partners of L. pneumophila str. Paris resistance proteins with high confidence value (0.7) were predicted. The protein sequences of resistance genes and their partners were retrieved from NCBI and STRING database. Finally, a comparative metabolic pathway was performed to analyze between L. pneumophila str. Paris and human as the host. All the proteins in the different pathways of L. pneumophila str. Paris and unique proteins of L. pneumophila str. Paris were collected in common pathways (between L. pneumophila str. Paris and human). All proteins in the different pathways of human and unique proteins of human in common pathways were excluded. The amino acid sequences of the selected proteins were downloaded from the Kyoto Encyclopedia of Genes and Genomes (KEGG) and NCBI. Total proteins of step I (Greenlist) were transferred to step II.
Step II
Host homology search
The main goal of this step is to find proteins in L. pneumophila str. Paris that have no similarity to the human proteome. This analysis minimizes the undesirable side effects of the drug.21,30 The Greenlist proteins were subjected to BLASTp against nonredundant protein sequences of human (taxid: 9606) with an e-value of 0.005.21,30,31 The Greenlist proteins that showed no hits for the 0.005 e-value were transferred to the next test (the test of essentiality).
Test of essentiality
The proteins in Greenlist were examined to recognize essential proteins using BLASTp search against DEG, a database of essential genes. The DEG BLASTp expected value cutoff was adjusted to 10−5. The hits with less than e-value of 10−5, identity ≥25%, and same annotated function of the query were selected as essential proteins.21,30,32 The proteins in Greenlist that revealed hits with DEG BLASTp parameters were collected and transferred to the next step (gastrointestinal flora homology test). The other proteins that revealed no hit in DEG BLASTp were excluded from the analysis.
Homology search for gastrointestinal flora
Unintentional targeting of vital molecules in the gut microorganisms may cause harmful effects on the host. To prevent this condition, a custom database was created from gut microflora reported in the literature and the proteins of Greenlist were subjected to BLASTp search against this custom database with an expected threshold of 10−4.17,20,21,32 The Greenlist proteins that showed >10 hits were excluded and the rest were selected as Whitelist. The Whitelist comprises the primary potential drug targets of L. pneumophila str. Paris.
Step III
Whole proteome comparisons
To find similarities, the proteome of important pathogens of the family Legionellaceae was compared. All proteomes belonging to these pathogens were compared in TaxPlot with a cutoff value of 200.
Homology search for Legionellaceae
To find homologues in important pathogens of the family Legionellaceae, primary potential drug targets of L. pneumophila str. Paris in Whitelist were subjected to BLASTp against a custom database. The BLASTp was done with an expected value of 10−4. Proteins with identity ≥50% were selected as final potential drug targets in the family Legionellaceae (Redlist) and were qualitatively characterized using various tools in step IV.
Step IV
The biological function of an unknown protein in the Redlist (hypothetical proteins) was determined by means of Pfam tool. Subcellular localization sites of some proteins were recognized in the literature, and web-based tools, such as Prediction of Subcellular Localization of bacterial proteins. (PSLpred), Prediction of Protein SORTing (PSORT), and subcellular localization predictor (CELLO), were used to predict possible subcellular localization sites of Redlist proteins. PSLpred is a web server that uses Support Vector Machines (SVMs) to predict protein subcellular localization. 33 PSORT is a computer program for the prediction of protein localization sites in cells. 34 CELLO is a web server for protein subcellular localization prediction and functional gene ontology analysis. 35 In the broad-spectrum search, a custom database of bacterial pathogens reported in the literature was created. 20 Proteins in Redlist were evaluated using BLASTp search against a custom database with an e-value of 0.005 for the identification of broad-spectrum targets. In the DrugBank database, 36 Redlist proteins were subjected to BLASTp search with an e-value of 10−5. The presence of molecule with the same biological function in BLAST results shows the druggability of the target molecules, and absence shows the novelty of the target molecules.21,32
Results
In this study, a subtractive homology-based method was used to identify a collection of potential therapeutic targets in some bacterial pathogens of the family Legionellaceae. A systematic pipeline that involved several computational tools and databases was developed (Fig. 1). Only these proteins were selected as acceptable drug targets that successfully passed the pipeline.
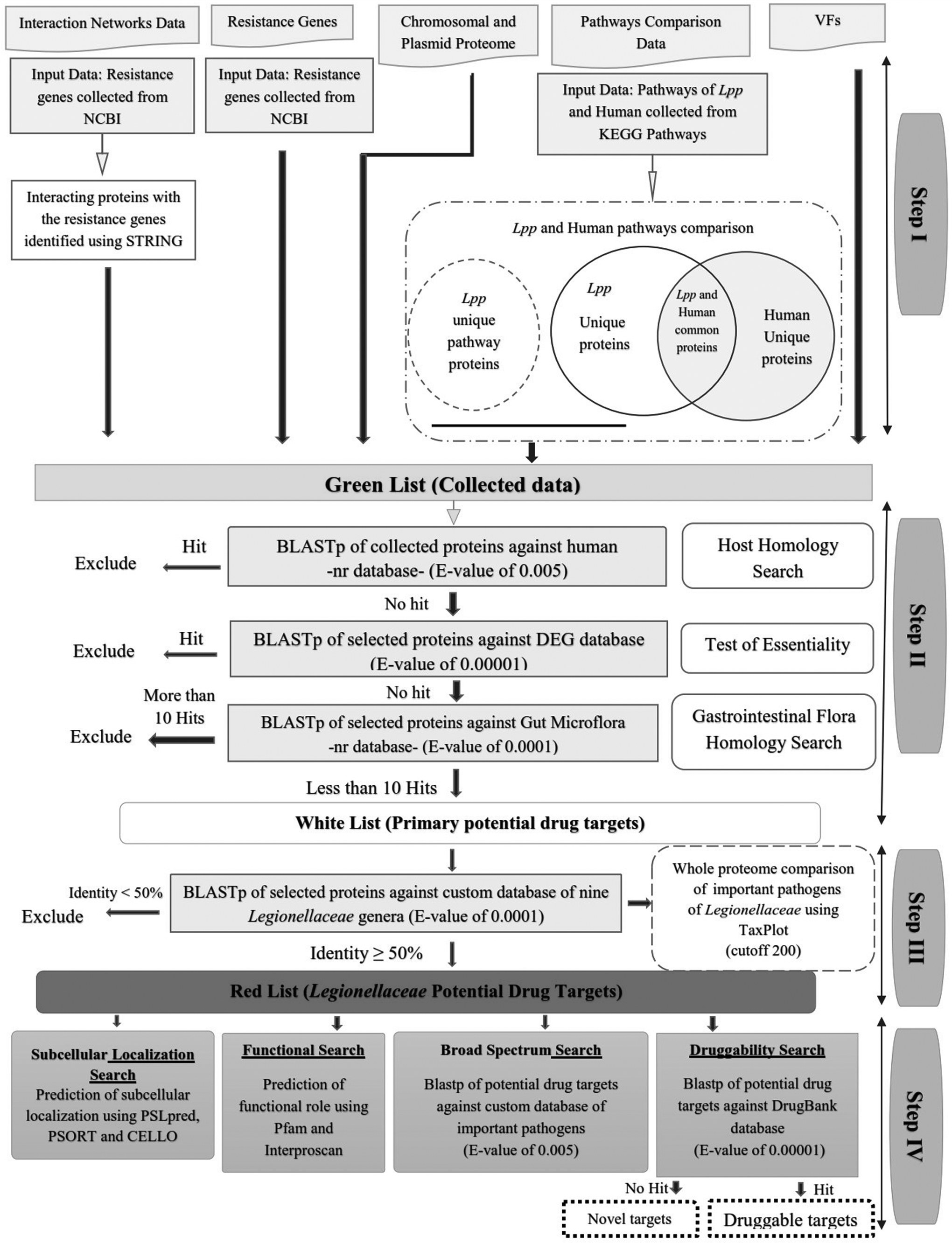
The complete flowchart of the homology-based method.
Step I
In the complete proteome (chromosomal and plasmid) of L. pneumophila str. Paris, 1,589 proteins were produced by positive strand and 1,557 proteins were translated from negative strand. Distributions of the protein lengths in the L. pneumophila str. Paris proteome were in the range of 44–7,679 amino acids. All 3,146 L. pneumophila str. Paris proteins were transferred to the next step. The major VFs in L. pneumophila str. Paris have been divided into the following seven groups: adherence, intracellular survival, invasion, iron acquisition, regulation, secretion systems, and toxins. All VFs (127 proteins) were transferred to step II. Inhibition of VFs proteins could reduce the virulence of pathogenic bacteria. Nine proteins that confer resistance to antimicrobial drugs in L. pneumophila str. Paris were obtained from the literature and corresponding sequences were retrieved from NCBI. Using STRING 10.0 tool and seven active prediction methods (neighborhood, gene fusion, co-occurrence, coexpression, experiments, databases, and text mining) with a high confidence value (0.7), 342 interaction partners for resistance-causing proteins were predicted. Antimicrobial resistance proteins and their interaction partners are considered as potential drug targets and suppression of these proteins may block the antimicrobial resistance process. 20 All the antimicrobial resistance proteins and their interaction partners (351 proteins) were transferred to the next step. In the KEGG database, there are 299 metabolic pathways for human and 109 metabolic pathways for L. pneumophila str. Paris. Pathways of L. pneumophila str. Paris are classified as metabolism, genetic data processing, environmental data processing, and cellular processes. Of 109 metabolic pathways in L. pneumophila str. Paris, 30 pathways were unique and 79 pathways were common with humans. Sequences of 531 unique proteins from common pathways and 203 proteins from distinct pathways were selected and transferred to step II. Finally, 4,358 proteins, including chromosomal and plasmid proteome, VFs, resistance proteins and their interaction partners, and common and unique metabolic pathway proteins were collected as Greenlist and transferred to step II.
Step II
A collection of 4,358 proteins that resulted from step I (Greenlist) were subjected to three sequential BLAST searches to find primary targets (Fig. 1). The first and third BLAST searches determined the proteins that were nonhomologous to human and gut microflora proteome. The essentiality of the proteins was assessed by second BLAST search against a known set of essential proteins in DEG database. Targeting of the proteins homologous to host by drug compounds could adversely alter the host metabolism. Therefore, filtering proteins homologous to human proteome is considered in several computational drug target discovery methods.17,21,30,32 A total of 4,358 L. pneumophila str. Paris proteins, including chromosomal and plasmid proteome (3,146 proteins), pathogenicity islands and VFs (127 proteins), resistance proteins and their interaction partners (351 proteins), and common and unique metabolic pathway proteins (734 proteins), were subjected to host homology search against the whole protein of the human. Proteins without any hits in BLAST search were considered as nonhomologous, whereas those showing hits were regarded as homologous. Of the 4,358 input proteins, 802 proteins that showed no hits against human proteome were selected and 3,556 proteins (homologous to human proteome) were excluded from the next step of the analysis. Essential genes are the genes of an organism that are vital for its survival. To preserve the vital process of an organism such as central metabolism, DNA replication, translation, cellular structure maintenance, and cellular transport, essential genes produce proteins. A drug target must be an essential protein for the survival of pathogenic microorganisms. Targeting of such proteins could cause growth inhibition and disrupt biological functions of microorganism. The Greenlist proteins were analyzed for determination of essentiality using DEG 11.3 database. Of 802 input proteins, 464 proteins were identified to be essential for the pathogen and selected for the next analyses. In addition, 338 proteins that showed no hits against DEG (nonessential) were excluded from the analysis. The gut microbiota plays an essential role in human metabolism, physiology, and nutrition, and it also plays an essential role in human health by preventing pathogens from colonization. Therefore, the decay of the gut microbiota population may cause problems in human health.37,38 The essential resulting proteins from previous analyses were subjected to BLASTp against a custom database of the whole proteome of gut microbiota. Of 464 input proteins, 60 proteins were collected as Whitelist and 404 proteins showing >10 hits were excluded. Whitelist proteins were selected as primary potential drug target of L. pneumophila str. Paris and were used for the next step of the analysis.
Step III
The reference proteome of L. pneumophila str. Paris was compared with the family Legionellaceae pathogens to find similarities. The Whitelist was subjected to BLASTp against a custom database of the family Legionellaceae pathogens from 60 proteins in the Whitelist. Eighteen proteins that showed identity ≥50% were selected as Redlist (Table 1). Redlist proteins were taken as the final potential drug targets in the family Legionellaceae (Table 2). The Redlist consists of proteins involved in metabolism, cellular transport, cell division, and cell motility. Proteins involved in metabolism and cellular transport provide a major contribution to the Redlist of final targets (13 of 18 proteins). Targets involved in multiple metabolic pathways are thought to be a more efficient drug target, and preventing the activity of such targets could increase lethal effects by blocking the activity of several metabolic pathways of the microorganism. Of the 18 Redlist targets, 8 targets (lpp0252, lpp0381, lpp1542, lpp1671, lpp1673, lpp2256, lpp2454, and lpp2758) were involved in >1 metabolic pathway (Table 2).
aa, amino acid; NCBI, National Center for Biotechnology Information; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Step IV
In this step, Redlist proteins were further explored using various analyses (functional search, subcellular localization search, broad-spectrum search, and druggability search) (Fig. 1). The biological function of unknown and hypothetical proteins in Redlist was determined by using Pfam tool. Eight proteins in Redlist, namely, lpp0375, lpp1542, lpp1720, lpp1729, lpp1975, lpp2256, lpp2683, and lpp2758 were unknown and hypothetical (Table 2). Hypothetical protein lpp0375 was determined to be an OstA-like protein. The OstA protein is known as an organic solvent tolerance protein. 39 Hypothetical protein lpp1542 was predicted to be a CDP-alcohol phosphatidyl transferase (CDP-AP). Phospholipids have key roles in the structure and function of all cell membranes. Members of CDP-alcohol phosphotransferase family are integral membrane proteins which are all involved in phospholipid biosynthesis. They catalyse the displacement of cytidine monophosphate (CMP) from a CDP-alcohol by a second alcohol with formation of a phosphodiester bond and concomitant breaking of a phosphoride anhydride bond. 40 Hypothetical protein lpp1720 was determined to be a flagellar FliJ protein, this family is found in bacterial motility proteins in flagellar system. Hypothetical protein lpp1729 was predicted to be an outer membrane (OM) lipoprotein carrier protein LolA that plays in the translocation of lipoproteins from the inner membrane (IM) to the OM. 41 Hypothetical protein lpp1975 was determined to be membrane-bound lytic transglycosylase (MltA)-specific insert domain. This beta barrel domain is found inserted into the MltA, a murein-degrading enzyme. This domain that contains three conserved aspartate residues may be involved in peptidoglycan binding. 42 Hypothetical protein lpp2256 was predicted to be a preprotein translocase subunit SecB. SecB is a cytoplasmic module common with membrane-bound multisubunit enzyme termed Sec protein translocase, which is the main component of the general secretory pathway (type II), involved in translocation of aborning polypeptides across the cytoplasmic membrane. 43 Hypothetical protein lpp2683 was determined to permease the YjgP/YjgQ family. Members of this family are integral membrane proteins of unknown function. Finally, hypothetical protein lpp2758 was predicted to be the cytochrome C1 family, which is the third complex in the electron transport chain. To identify cellular localization, Redlist proteins were analyzed using PSORT, PSLpred, and CELLO tools. In PSORTb and PSLpred, based on the localization score, reliability index, and expected accuracy, five targets were predicted to be cytoplasmic proteins, five targets were predicted to be cytoplasmic membrane proteins, two targets were predicted to be periplasmic, and two targets were predicted to be the OM protein. The cellular localization of four targets (lpp0252, lpp0922, lpp1673, and lpp2664) was predicted to be unknown. Furthermore, to obtain greater accuracy, the predicted subcellular localization of the targets was cross-evaluated using the CELLO tool. In CELLO, the location of six targets was determined as cytoplasmic, the location of two targets was determined as periplasmic, the location of nine targets was determined as IM, and the location of one target was determined as OM. Of the four targets characterized as unknown by PSORTb and PSLpred, one target was predicted as cytoplasmic, one target was predicted as periplasmic, and two targets were predicted as IM (Table 3). Six proteins with cytoplasmic localization and nine proteins with IM localization and one protein with OM localization could, respectively, possibly serve as drug and vaccine targets (Table 3). In a broad-spectrum search, BLASTp against a custom database of the whole proteome of bacterial pathogens resulted in the recognition of broad-spectrum targets. Results of the broad-spectrum search showed that 7, 8, and 3 targets were present in >100 pathogens, between 50 and 100 pathogens, and <50 pathogens, respectively. Seven proteins of Redlist that were present in >100 pathogens were considered as broad-spectrum targets (Table 3). Drug targets involved in vital metabolic process seem to be a broader spectrum than the other targets such as VFs. VFs are not often broad-spectrum drug targets because of high specificity. Targeting of broad-spectrum proteins by drug molecules may facilitate the destruction of a wide range of pathogenic bacteria. One target that is present in <25 pathogens was considered as the family Legionellaceae-specific targets. Such specific target proteins may decrease the threat of antimicrobial resistance development in a wide range of pathogenic bacteria. 20 Druggability search was the final analysis in this approach. Druggability of Redlist targets was assessed using BLASTp against DrugBank database. Similarity search showed that four targets (lpp0252, lpp1671, lpp1673, and lpp2454) are homologous to each other or more known targets in DrugBank database with an expected value of 10−5. BLASTp against DrugBank also showed that the lpp0252 target has three experimental inhibitors [2-amino-3-(1-hydroperoxy-1 h-indol-3-yl) propan-1-ol, ethionamide, and isoniazid], lpp1671 has one experimental inhibitor (formic acid), lpp1673 has two experimental inhibitors (N ∼ 2∼-succinylarginine and N ∼ 2∼-succinylornithine), and lpp2454 has three experimental inhibitors [2-amino-3-(1-hydroperoxy-1 h-indol-3-yl) propan-1-ol, ethionamide, and isoniazid]. All these four druggable targets are involved in vital pathways of amino acid metabolism of L. pneumophila str. Paris. Fourteen targets, showing no homology with DrugBank database, were considered as novel drug targets, which should be further evaluated experimentally (Table 3).
MltA, membrane-bound lytic transglycosylase; CDP, cytidine diphosphate.
Discussion
Recent advances in whole genome sequencing and computational methods have facilitated the discovery of novel antimicrobial agents and could enable us to combat antibiotic resistance in bacteria. In addition, they have provided important insights into the molecular mechanisms of underlying diseases. The development of new bioinformatic approaches and modulation and optimization of existing strategies can increase the accuracy of drug target identification. 44 This study has shown how bioinformatics and in silico approaches can be used in the drug discovery processes. As biological systems are complex networks of many metabolic and nonmetabolic pathways, the explanation of such systems may be obtained by considering large-scale studies. Focusing on a single drug target at a time in conventional drug discovery methods may not always produce acceptable or expected results. Comprehensive study of biological pathways and whole cell systems provides wider insights into the fitness of a potential drug target. Some proteins in an organism that may be predicted as good drug targets, when viewed in the context of a biological system, may not actually be vital. Analyzing drug targets in the context of a biological system can help in evaluating the criticality of the individual targets in the cell pathways. Of 4,358 proteins analyzed, 18 (Redlist) were selected as final putative drug targets in the family Legionellaceae (Table 2). Putative targets were filtered to exclude nonviable candidates based mostly on importance of survival, lack of homology to the human host, well-known biological function, and conserved between Legionellaceae species. The Redlist consists of proteins involved in metabolism (amino acid, energy, and lipid metabolisms), cellular transport, cell division, and cell motility. These essential genes that play a great role in cell survival encode the proteins to control a central metabolism, replicate DNA, translate genes into proteins, maintain a basic cellular structure, and mediate transport processes within or out of the cell. 45 The essential proteins are involved in many biological pathways that represent molecular interaction networks between the pathogen and the host. Essentiality search shows some vital proteins that are required by the pathogen to perform important roles for their survival, growth, and replication. Therefore, a broader search of bioinformatics is a critical stage to predict potential drug target in the pathogens. Proteins involved in metabolism and cellular transport provide a major contribution to the Redlist of final targets (14 of 18 proteins). Of the 18 Redlist protein targets, 8 targets (lpp0252, lpp1542, lpp1671, lpp1673, lpp2256, lpp2454, lpp2758, and lpp2973) are involved in >1 metabolic pathway (Table 2). Restricted targeting of some specific pathways may cause development of multidrug resistance among pathogenic bacteria. 21 In general, approaches that consider all essential pathways of organisms can be more successful to identify efficient drug targets. Targets involved in multiple metabolic pathways are thought to be a more efficient drug target, and preventing the activity of such targets could increase lethal effects by blocking the activity of several metabolic pathways of the microorganism. Targeting of metabolic enzymes for cancer therapy is a hot topic for drug discovery. 46 Molecular structures of membrane transporter proteins are significant for drug discovery. These proteins are important coplayers in cellular systems and are known molecular components of many disease processes. The membrane transporter proteins are targeted by many currently used drugs and have a major potential as targets for new drug development. A large number (60–70%) of the current known drug targets are proteins set in a cellular membrane, and membrane proteins are among the most interesting macromolecules to study using structural biology techniques. High-resolution structural knowledge about proteins placed in a cellular membrane is not only of pivotal importance for developing new drugs with therapeutic potential, but is also necessary for the knowledge of the molecular mechanisms of cellular signaling and function. 47 The lpp2758 target is involved in the bacterial two-component system. By using the two-component system, the pathogens perceive the changes in the environment and respond to it. Moreover, involvement in the pathogenicity of the organisms and the absence of these systems in humans make them attractive drug targets. 48 Therefore, inhibition of these proteins can reduce the growth rate and virulence of the pathogen. Targeting of proteins involved in DNA replication, DNA repair, DNA recombination, and cell division (lpp2664 and lpp1690) may disrupt the pathways essential for pathogen survival, growth, and reproduction. Seven broad-spectrum targets are involved in crucial processes such as cellular transport, environmental information processing, and metabolism (Tables 2 and 3). Drug targets involved in the vital metabolic process seem to have a broader spectrum than the other targets such as VFs. Because of high specificity, VFs are not often broad-spectrum drug targets. Targeting of broad-spectrum proteins by drug molecules may facilitate the destruction of a wide range of pathogenic bacteria. Eleven Legionellaceae-specific targets are suitable for the development of narrow-spectrum antibiotic. Such specific target proteins may decrease the threat of development of antimicrobial resistance in a wide range of pathogenic bacteria. Based on the localization analysis, most targeting proteins were located in the membrane (Table 3). Inhibition of target proteins located on the membrane and extracellular proteins is important because of their crucial role as VFs assisting pathogens to spread and proliferate within the host. Knowledge of protein localization is valuable for learning the function and the interaction of different proteins. When other information is not available, the subcellular localization will also be effective in the annotation for new proteins. In drug discovery process, subcellular localization data can help understand therapeutic intervention points. For example, because of their localization, secreted proteins and membrane proteins are easily available by drug molecules. 49 The Gram-negative bacteria are surrounded by two membranous structures, the IM and the OM. The IM that is called the plasma membrane has a trilamellar structure surrounded with bacterial protoplasm and composed of a phospholipids bilayer. Most of the membrane proteins that have role in energy production, lipid biosynthesis, protein secretion, and transport system are conserved in bacteria, but their cellular location is different. In bacteria, these proteins are located in the IM.50,51 The OM also presents a trilamellar structure (with couple electron dense leaflets, outer and inner) in the electron micrograph and consists of proteins, containing porins, receptors, and an asymmetric distribution of lipids. 50 The OM of Gram-negative bacteria provides a difficult barrier that must be overcome. There are essentially two pathways that antibiotics can take within the OM: a lipid-mediated pathway for hydrophobic antibiotics and general dissemination porins for hydrophilic antibiotics, which consequently may lead to antibiotic resistance. 52 Druggability refers to the ability of a target molecule to bind with high affinity to the drug molecules. Druggability is one of the most important characteristics of a target molecule. Four druggable targets (lpp0252, lpp1671, lpp1673, and lpp2454) are involved in vital process such as oxidant–antioxidant system, metabolism, and biosynthesis of macromolecule. Either of these proteins is a target for conventional antibiotics. Fourteen novel drug targets are suitable for development of new antimicrobials and should be further evaluated experimentally. Currently, several computational methods such as comparative genomics, data mining, structure and sequence to function, and metabolic pathways are used for the identification and characterization of the drug targets. Most of these target discovery methods enable us to identify potential drug target candidates based on the main criteria of specificity and essentiality. Besides these properties, a suitable drug target must be specific to the pathogen, avoid harmful side effect, and should be a very important protein for survival of the pathogen. Suppression of such drug targets can result in effective control of the pathogen without any harmful effects on the host. Our homology-based method considers the essentiality, specificity, druggability, subcellular localization, function, and broad-spectrum condition of drug targets. Although a sequence similarity of the protein does not ensure the same structures or binding properties, using such homology-based methods could ease the optimization and production of new drugs and vaccines. In conclusion, the homology-based method17,20,21,32 was applied to identify novel putative drug targets in the family Legionellaceae. The results of this study identified several proteins in the genomes of the family Legionellaceae pathogens that can be targeted for effective drug design and development. Thus, many of these putative drug targets that are involved in several vital metabolic pathways, such as energy metabolism, amino acid metabolism, and lipid metabolism, designing drug molecules against these targets could be very effective for the treatment of Legionella infections. Development of new drug against such targets will be precise to the pathogen and considerably decrease the harmful side effects to the host. The efficiency of already available antimicrobial drugs can be tested using this method. Targeting of proteins involving several essential metabolic pathways may facilitate the efficient treatment of infections. The findings of such studies facilitate the design and development of novel antimicrobial drugs against the family Legionellaceae and other pathogens.
Footnotes
Acknowledgments
The authors would like to thank all staff of the Bioinformatics Lab, School of Medicine, Kerman University of Medical Sciences, for their kind cooperation.
Disclosure Statement
No competing financial interests exist.