
Abstract
Fluoroquinolones are the drug of choice for most of the infections caused by Escherichia coli, and their indiscriminate use has resulted in increased selective pressure for antibiotic resistance. At present, sequencing is the only reliable and direct technique to detect mutations in the quinolone resistance determining region (QRDR). In this study, a rapid and reliable mismatch amplification mutation assay (MAMA) PCR to detect mutations in the QRDR was evaluated and compared to PCR-restriction fragment length polymorphism (PCR-RFLP). One hundred one clinical isolates of E. coli were subjected to MAMA-PCR and PCR-RFLP to detect QRDR mutations. Overall, 92 (91.08%) resistant isolates harbored a point mutation of S83L in gyrA. Double mutations in gyrA were also detected in 45 (44.55%) isolates. Similarly, 41 (40.59%) isolates possessed a point mutation at parC 80, and 25 (24.75%) isolates possessed a point mutation at parC 84. Additionally, MAMA-PCR—the first of its kind—was also standardized to detect mutations in regions gyrB 447 and parE 416, although no mutations were detected in these regions. The rapid and sensitive MAMA-PCR method evaluated in this study would be helpful in exploring the underlying mechanism of fluoroquinolone resistance to enhance control strategies.
Introduction
E
Such E. coli strains also result in multidrug resistance, which is a major concern for global public health. Thus, fighting antibiotic resistance in Gram-negative bacteria calls for a sturdy, more nuanced, and better coordinated approach. It is therefore essential to study the resistance mechanisms in pathogenic E. coli for the development of suitable therapeutic strategies. Mutations in the bacterial DNA gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE), presence of plasmid-mediated quinolone resistance (PMQR) genes and overexpression of active efflux pumps are the known fluoroquinolone resistance mechanisms.5,6 Among these, mutations in the quinolone resistance determining region (QRDR) are the most frequently observed phenomenon in resistant strains. 7
Topoisomerases are known to play an important role in DNA replication, where DNA gyrase or topoisomerase II (two GyrA and two GyrB subunits) introduces negative supercoiling into DNA, and topoisomerase IV (two ParC and 2 ParE subunits) is responsible for removing the interlinking structure between the two newly produced DNA strands during replication. Fluoroquinolones are the synthetic class of antibacterial agent that act on both DNA gyrase and topoisomerase IV and have many clinical applications. Quinolone and fluoroquinolone usually differ in their activity for DNA gyrase and topoisomerase IV. The mutations in the QRDR inhibit the binding of fluoroquinolone to DNA and the enzyme (topoisomerase targets) complex. 8 QRDR mutations and their association with the fluoroquinolone resistance in E. coli have been well studied by DNA sequence analysis of resistant strains. 9
Several point mutations have been found to be responsible for resistance in E. coli. These mutations are found most frequently in the Ser 83 and Asp 87 codons of the gyrA in E. coli, as well as in other microorganisms. 10 Over the years, several methods have been developed to detect such mutations.11,12 However, the sequencing technique is the most appropriate method of identifying mutations in any gene, but it has a high cost and is labor intensive and time-consuming. PCR-restriction fragment length polymorphism (PCR-RFLP) is another technique that has been utilized extensively in the detection of certain disease-causing genetic mutations. 13 However, its ability to detect all known QRDR mutations is not clearly understood.
To overcome the shortcomings of high sequencing costs, this study evaluated the simple, rapid, time-efficient mismatch amplification mutation assay-PCR (MAMA-PCR) method developed by Qiang et al. 14 for the detection of QRDR mutations and also compared it with PCR-RFLP to confirm the point mutations in the QRDR (gyrA, gyrB, parC, and parE) of E. coli. MAMA-PCR primers are designed in such a way as to create a double mismatch with the mutated genes but a single mismatch with the wild-type gene. As this method is sensitive and specific in identifying the point mutations, this technique could be profitably applied as a standard genetic screening formula.
Materials and Methods
Bacterial strains and determination of antimicrobial susceptibility testing
One hundred one E. coli isolates from clinical samples (UTI) were revived from 30% glycerol stock in Luria Bertani Broth (HiMedia Laboratories, Mumbai, India) and subjected to antibiotic susceptibility testing. The isolates were analyzed for the uinolone and fluoroquinolone susceptibility using a disc diffusion assay according to Clinical and Laboratory Standards Institute (CLSI) guidelines. 15 Briefly, the bacterial isolates with a density of 0.5 McFarland turbidity were swabbed onto pre-poured and dried Mueller–Hinton agar. The antibiotic discs of ciprofloxacin (5 μg) and NA (30 μg; HiMedia Laboratories) were placed on the bacterial lawn using a sterile applicator. After overnight incubation at 37°C, inhibition zone diameters were measured and interpreted as resistant, sensitive, or intermediate, as per the CLSI guidelines. 15 E. coli (ATCC 25922) was used as a quality control strain.
Detection of mutation in the QRDR by MAMA-PCR
MAMA-PCR 14 was performed for all 101 isolates to detect the presence of mutations in the QRDR. The known mutations at amino acid positions 83 and 87 of gyrA, amino acid positions 80 and 84 of parC, amino acid position 447 of gyrB, and amino acid position 416 of parE were targeted. The primers for gyrB and parE were designed using the Primer3 bioinformatics tool with the introduction of one mismatch at 3′ ends of the reverse primer. The primers used in the study are listed in Table 1.
Oligonucleotide Primers Used for Determining Gyrase and Topoisomerase IV Target Genes
MAMA, mismatch amplification mutation assay.
DNA was extracted using the method described by Ausubel et al. 16 PCR was performed in 30 μl volumes containing 3 μl of 10× Taq buffer, 83 μM of each of the four deoxyribonucleotide triphosphates, 30 pmol of forward primer, 20 pmol of MAMA reverse primer, 10 pmol control reverse primer, and 1.0 IU of Taq DNA polymerase with 2 μl of template DNA. Amplification was carried out using a thermal cycler (Bio-Rad Laboratories, Hercules, CA) with initial denaturation at 95°C for 5 min and 35 cycles of denaturation at 94°C, annealing at 55°C, and extension at 72°C, each for 40 s, followed by a final extension of 72°C for 10 min. PCR products were visualized on 2% agarose gel pre-stained with ethidium bromide (0.5 μg/ml) in 1× TAE buffer loaded with 10 μl of the reaction mixture and observed under UV light in a gel documentation system (Bio-Rad).
PCR-RFLP for the detection of mutations in gyrA and parC
To confirm and compare the results of MAMA-PCR, PCR-RFLP was performed for all the isolates. The target restriction sites (gyrA 83 and parC 80) were digested using two different restriction enzymes: HinfI and EcoRV, respectively.
PCR was performed in 30 μl volumes containing 3 μl of 10× Taq buffer, 50 μM concentrations each of dNTPs, 10 pmol of each of the forward and reverse primers (for gyrA and parC), and 1.0 IU of Taq DNA polymerase (HiMedia Laboratories) with 2.0 μl of template DNA. Amplification reactions were carried out as discussed earlier using a thermal cycler (Bio-Rad) with an annealing temperature of 55°C for 30 s. The obtained gyrA PCR products were digested with HinfI (Fermentas; Thermo Fisher Scientific [India], Mumbai, India) to screen for point mutations at position Ser-83, and parC PCR products were digested with EcoRV (Fermentas; Thermo Fisher Scientific [India]) to screen for point mutation at Ser-80. The enzymes used and their restriction sites are listed in Table 2. Enzyme digestion was performed in a 20 μl mixture containing 16 μl (0.1–0.5 μg) of the PCR product, 0.5 μl (2 IU) of enzyme, 2 μl of 10× buffer, and 1.5 μl of sterile ultrapure water at 37°C for 2 h followed by termination of enzyme activity at 65°C for 10 min using a dry bath (GeNei™; Merck Specialities, Hyderabad, India). The digested PCR products were electrophoresed as discussed earlier and visualized under UV light in a gel documentation system (Bio-Rad).
Restriction Enzymes with Their Restriction sites
DNA sequencing
PCR products of two representative isolates were outsourced for sequencing (Eurofins Genomics India, Bangalore, India), and the generated sequences were analyzed using the NCBI's BLAST program (http://blast.ncbi.nlm.nih.gov). The corresponding amino acid sequences were obtained from a molecular tool kit, and the sequences were submitted to GenBank.
Results
Antibiogram analysis
All 101 E. coli isolates harboring uidA (genus specific gene) were checked for quinolone and fluoroquinolone resistance using nalidixic acid and ciprofloxacin antibiotic discs. Overall, 95 (94.05%) isolates were found to be resistant, and six (5.94%) were sensitive to the antibiotics. Among the resistant isolates, 13 (12.87%) showed resistance only to nalidixic acid, while the remaining 88 (87.12%) isolates exhibited resistance to both nalidixic acid and ciprofloxacin.
Detection of mutations in the QRDR using MAMA-PCR
In MAMA PCR, in the absence of mutation(s) in sensitive isolates, two PCR products were generated from the wild-type gene using universal forward/reverse and MAMA reverse primers. On the contrary, PCR was inhibited due to two or more mismatches at the 3′ end of the MAMA primer in the presence of mutation(s), and single amplification was observed in resistant isolates (Fig. 1A, C, E, F). Thus, the MAMA primers were able to distinguish sensitive and resistant strains used in this study. Twenty (19.80%) of the 101 E. coli isolates were found to harbor a mutation only at amino acid position 83 of gyrA, and one of the isolate (S5) had a mutation only at position 87 of gyrA. Twenty-seven (26.73%) resistant isolates harbored mutations in gyrA 83 and 87 and parC 80 regions. Three of the isolates that were only resistant to nalidixic acid possessed mutations at gyrA 83 and 87 and parC 80. Ten (9.90%) resistant isolates had mutations at amino acid position 83 of gyrA and 80 of parC, and 17 (16.83%) isolates showed mutations at position 83 of gyrA and 84 of parC. Four isolates (3.96%) of E. coli resistant to both the antibiotics found to display mutations at all four important positions in the QRDR (gyrA 83 and 87 and parC 80 and 84) and two isolates (1.98%) resistant to nalidixic acid did not harbor any mutations in the QRDR (Table 3). None of the isolates showed mutations in suspected gyrB and parE regions. The PCR products of gyrA and parC from the two representative isolates of E. coli were sequenced and analyzed. The partial sequences with possible QRDR mutations were submitted to GenBank and assigned GenBank accession numbers (gyrA: MF288967; parC: MF2889868).
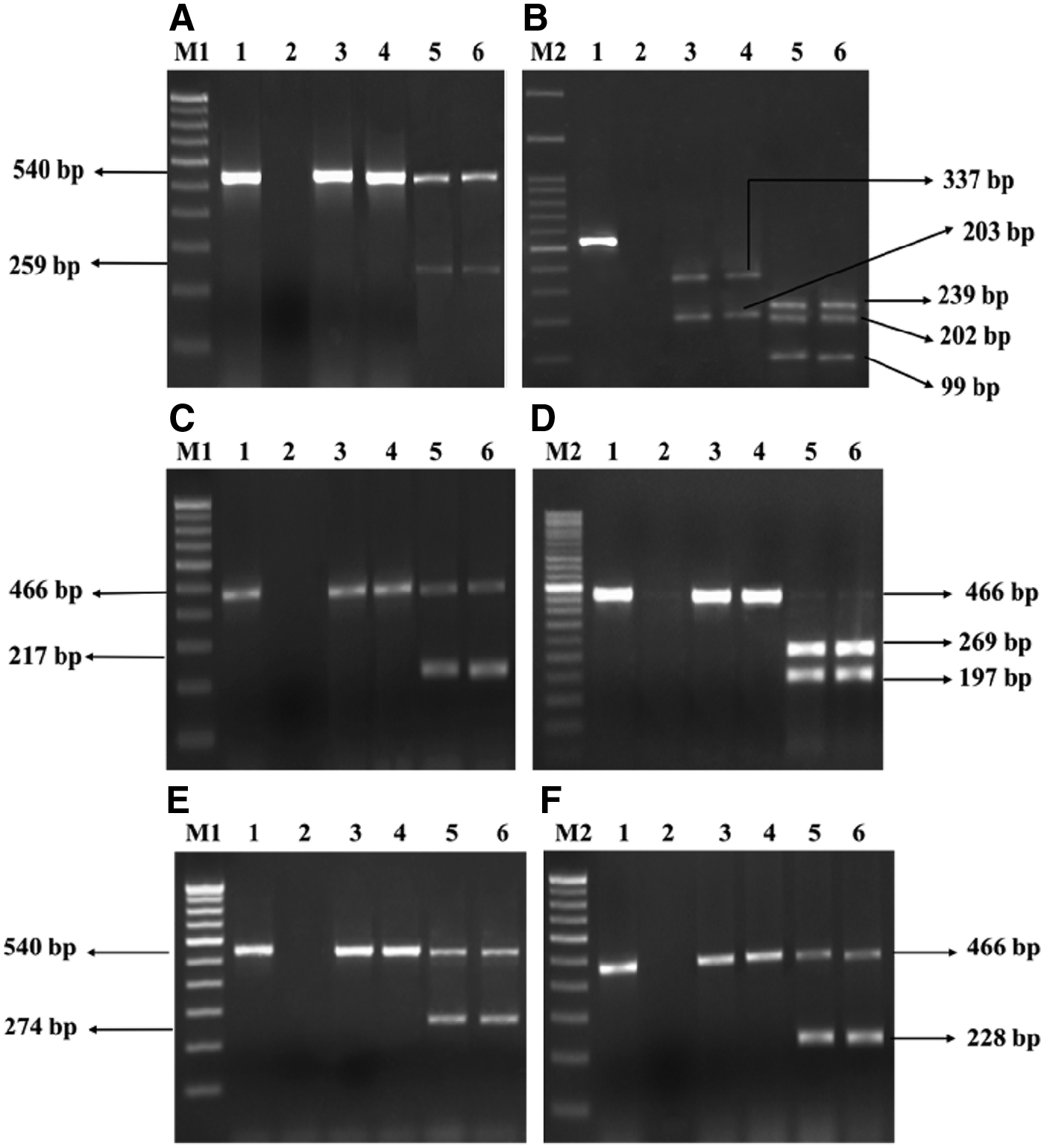
Agarose gel electrophoresis image of PCR products generated from duplex MAMA-PCR assays and PCR-RFLP.
Mutation Status of Escherichia coli Isolates at Topoisomerase Targets
Isolates sensitive to both the antibiotics.
Nalidixic acid–resistant and ciprofloxacin-sensitive isolate with no mutations at QRDR.
QRDR, quinolone resistance determining region.
Detection of mutations in the QRDR using RFLP
MAMA-PCR products of the 101 isolates were subjected to PCR-RFLP to detect mutations in the QRDR. In the case of gyrA PCR-RFLP, the restriction enzyme HinfI produced two bands for resistant isolates due to the abolishment of the restriction site at position 83, which was found in sensitive isolates, resulting in three bands (Fig. 1B) (Table 4). The mutation in the parC region of resistant isolates resulted in the formation of a restriction site at position 80 for EcoRV and thus generated two bands, whereas a single band was observed in the susceptible isolates due to the absence of restriction sites for EcoRV in parC (Fig. 1D; Table 4). The isolates with a point mutation in the QRDR detected using MAMA-PCR also produced similar results with PCR-RFLP in gyrA 83 and parC 80 regions. MAMA-PCR showed 100% categorical agreement with the PCR-RFLP in detecting mutations in gyrA 83 and gyrA 87 regions. However, irrespective of whether the isolates are sensitive or resistant to fluoroquinolones, the PCR-RFLP was inapplicable for detecting mutation in gyrA 87 and parC 84 regions due to the absence of restriction sites for any known restriction enzymes. Further, two representative resistant isolates (S20 and J5) with a mutation in gyrA and parC regions were selected for sequencing to confirm the mutation. Isolate S20 harbored a double mutation with amino acid substitution at 83 and 87 amino acid positions of gyrA. In gyrA 83, serine (TCG) was changed to leucine (TTG) due to a single nucleotide mismatch/point mutation, and in gyrA 87, aspartic acid (GAC) was changed to asparagine (AAC). The isolate S20 also harbored a single point mutation at parC 80, with a change of serine (AGC) to isoleucine (ATC). A double mutation was observed in both gyrA and parC of isolate J5, with a change of serine (TCG) to leucine (TTG) at gyrA 83 and a change of aspartic acid (GAC) to asparagine (AAC) at gyrA 87 (Fig. 2A). Similarly, serine (AGC) was changed to isoleucine (ATT) at parC 80, and glutamic acid (GAA) was changed to valine (GTA) at parC 84 (Fig. 2B) in J5. The sequencing results of these two isolates are in congruence with the results of MAMA-PCR.
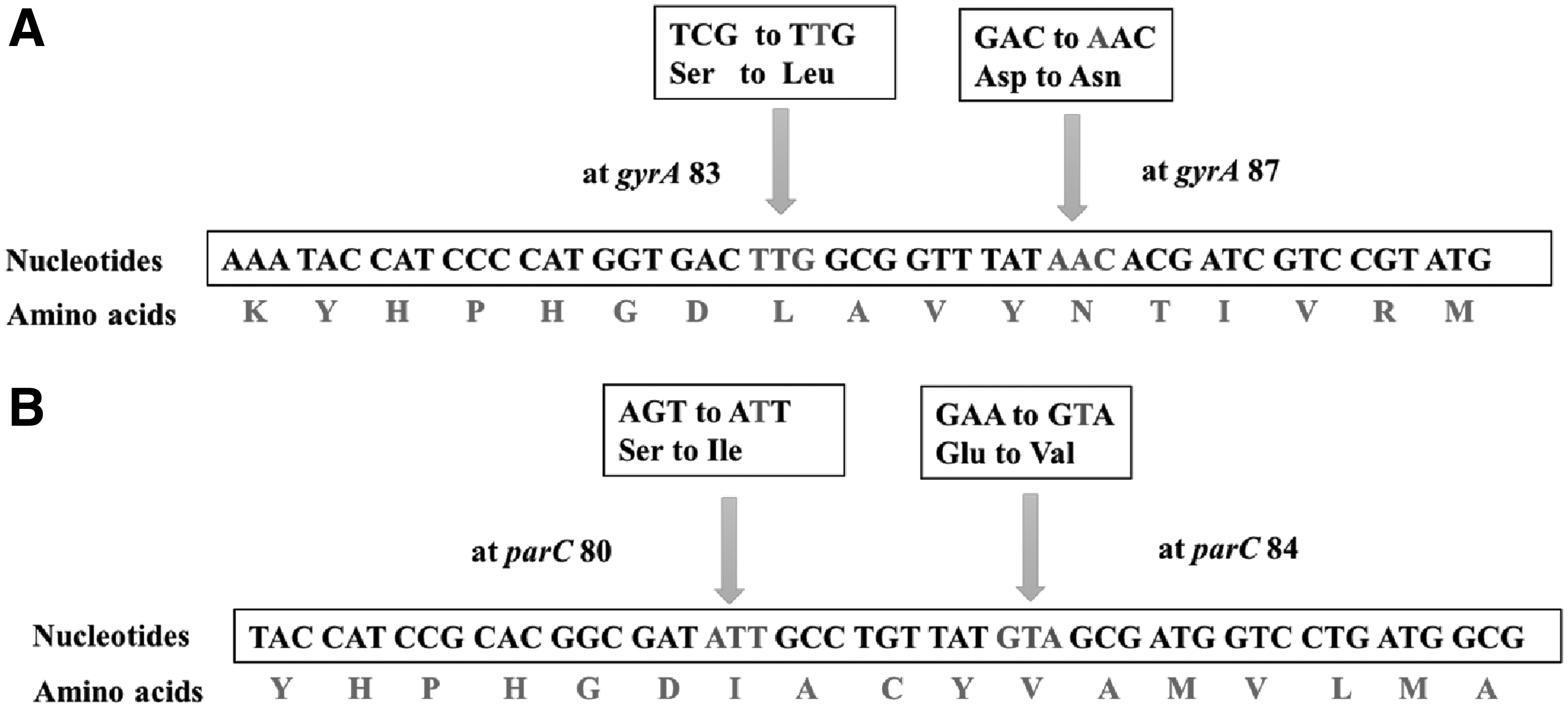
Quinolone Resistance Determining Region Mutation Pattern and Restriction Fragment Length Polymorphism Results of Escherichia coli Isolates
A, mutation absent; P, mutation present.
Discussion
Microorganisms exhibit varied resistance mechanisms to overcome the action of antimicrobial agents such as fluoroquinolones. To develop feasible therapeutic strategies to treat the infections caused by E. coli, it is important to understand the mechanisms of resistance in these pathogens. Studies on the fluoroquinolone resistance mechanism caused by mutation in the QRDR in Gram-negative bacterial pathogens such as E. coli 18 and Salmonella 19 have been well documented. Although several molecular techniques are available to detect these mutations, including sequencing, RFLP, single-strand conformation polymorphism, and multiplex single nucleotide polymorphism typing assay, it is important to develop a rapid, sensitive, and cost-effective method. Therefore, MAMA-PCR was evaluated in this study. This technique utilizes duplex reverse primers along with the forward primer to detect mutations in the QRDR. In MAMA-PCR, a single nucleotide mismatch at the 3′ end of reverse primer causes Taq DNA polymerase to be unable to extend the primer.10,20 Thus, amplification will be inhibited if there is an additional mutation in the gene. Although many studies have used MAMA-PCR for the detection of QRDR mutations in resistant bacterial pathogens,14,21,22 there is a paucity of data on the efficiency of MAMA-PCR and PCR-RFLP in the detection of QRDR mutations.
MAMA-PCR developed to detect the point mutations in the QRDRs of fluoroquinolone-resistant isolates of E. coli was found to be highly efficient in detecting mutations in the four major QRDRs gyrA 83, gyrA 87, parC 80, and parC 84. The study is in agreement with the earlier reports, where MAMA-PCR was successfully utilized to detect mutations in gyrA and parC regions in fluoroquinolone-resistant bacterial pathogens.14,21–25 The point mutation in gyrA 83, S83L, was the most commonly observed change26–29 followed by the mutation in gyrA 87, which is in accordance with the present study, with 91.08% of isolates having a point mutation in gyrA 83 and 45.54% in gyrA 87. Considering other important mutations that could influence the level of resistance, MAMA-PCR was standardized to detect the mutations in regions gyrB 447 and parE 416, but none of the E. coli isolates used in the study had mutations in these regions. However, it is important to analyze these regions to check the multiple mutation status of resistant pathogens.
Among all 101 isolates of E. coli, eight different patterns of QRDR mutations were observed (Table 3). The majority of the fluoroquinolone-resistant isolates harbored mutations in three highly prevalent QRDRs: gyrA 83, gyrA 87, and parC 80. Most of the isolates that harbored a mutation in gyrA 87 were found together with the gyrA 83 mutation. However, one (S5) nalidixic acid–resistant isolate was found to harbor a mutation only in gyrA 87. Higher levels of quinolone/fluoroquinolone resistance in many bacterial isolates are associated with double mutations in the gyrA region. 30 Similarly, in this study, the isolates that had double a mutation in gyrA and other regions were resistant to both nalidixic acid and ciprofloxacin. Compared to the rate of mutations in gyrA 83 and 87, the percentage of mutations in parC 80 and 84 were less than gyrA mutations in an earlier report, 30 which was also seen in this study. Surprisingly, an isolate resistant to nalidixic acid did not harbor any mutation in the QRDRs. This strengthens the earlier report of fluoroquinolone-resistant isolates harboring PMQR genes or genes coding for efflux pumps as an additional mode of resistance mechanism. 18
To compare the results of MAMA-PCR, PCR-RFLP was also performed for all 101 E. coli isolates. Many reports have suggested the use of PCR-RFLP as an alternative for gene sequencing to detect mutations in the QRDR.31–33 Although both of the enzymes used in this study could detect mutations in gyrA 83 and parC 80 regions, there are no restriction enzymes available to detect mutations in gyrA 87 and parC 84 regions. It is one of the advantages of MAMA-PCR over PCR-RFLP that it can detect the mutations even in other mutation-prone regions using MAMA primers, which are designed exclusively to detect these mutations.
In addition to this, four isolates resistant to both nalidixic acid and ciprofloxacin showed a mutation in all the suspected sites (gyrA 83, gyrA 87, parC 80, and parC 84) by MAMA-PCR, but PCR-RFLP failed to detect the mutations at gyrA 87 and parC 80 due to the absence of a restriction site, even though there was a mutation. The results of MAMA-PCR and PCR-RFLP in detecting mutations in the QRDR of these isolates created enough ambiguity to proceed to sequencing, which is the ultimate technique to analyze the nucleotide sequence change. The DNA sequencing of gyrA and parC of one of the isolates (J5) revealed changes S83L and D87N in gyrA (Fig. 2A) and I80S and E84V in parC (Fig. 2B), and it further confirmed the results of MAMA-PCR. The fidelity of MAMA-PCR in detecting the above-stated mutation revealed its potential over PCR-RFLP to detect mutations at all the major sites of QRDR. Thus, the technique is efficient in detecting single nucleotide changes in all the major QRDRs compared to PCR-RFLP. However, the MAMA-PCR used in this study could only detect the presence of mutations in the QRDRs rather than detecting the type of amino acid change. Further, the technique can only be used effectively for the detection of known point mutations, whereas sequencing of QRDR always offers a better alternative to detect any silent or new novel point mutations responsible for fluoroquinolone resistance. Nevertheless, MAMA-PCR was found to be an ideal technique when a large number of drug-resistant bacterial pathogens need to be screened for epidemiological investigations.
In conclusion, the MAMA-PCR assay developed in this study was found to be more useful than PCR-RFLP in detecting QRDR mutations. Although RFLP could detect the mutations at gyrA 83 and parC 80, no restriction sites are generated or abolished if the isolates have novel point mutations in regions other than gyrA 83 and parC 80. Hence, RFLP cannot be used as a sole technique if the isolates have mutations in regions gyrA 87 or parC 84. Since multiple mutations in the QRDR are a major contributor to high-level fluoroquinolone resistance by reducing the minimal inhibitory concentration level in Gram-negative isolates, a MAMA-PCR assay was also developed for the first time in the present study to detect mutations in regions gyrB and parE of the QRDR. It is important to be vigilant about the antimicrobial resistance/sensitivity pattern and mechanisms of resistance of human pathogens such as E. coli to develop new therapeutic strategies and to prevent treatment failures. Therefore, we look in favor of controlling antibiotic abuse and encourage the use of different molecular techniques to detect different mechanisms of antimicrobial resistance acquired by human pathogens.
Footnotes
Acknowledgments
Financial support received by D.V.K. from the Government of India funded DST-SERB (Grant no. ECR/2017/000559) and Nitte University toward this study is gratefully acknowledged.
Disclosure Statement
No competing financial interests exist.