
Abstract
Resistance to aminoglycoside antibiotics is now common in pathogenic bacteria, making treatment of infections difficult. The rapid spread of resistance is mainly related to the dissemination of genes encoding aminoglycoside-modifying enzymes (AMEs). Staphylococci and enterococci are opportunistic human pathogens capable of causing a wide range of infections. Isolates from clinical cases are often found to be resistant to aminoglycosides. The aim of the present study was to develop a bead-based xTAG assay for the simultaneous detection of five prevalent aminoglycoside resistance genes in staphylococci and enterococci, including aac(6′)-Ie-aph(2″)-Ia, aph(3′)-IIIa, ant(4′)-Ia, ant(9)-Ia, and ant(6)-Ia. The limit of detection ranged from 10 to 1000 copies/μL of input purified plasmid DNA. Twenty-two bacterial isolates from clinical samples were examined using the newly developed xTAG assay and also by conventional PCR to determine the relative performance of each. The results obtained by xTAG assay showed higher detection rates and accuracy for AME genes than conventional PCR. It indicated that the xTAG-multiplex PCR method is a high-throughput tool for rapid identification of AME genes.
Introduction
Staphylococcus aureus and Enterococcus species are significant opportunistic human pathogens capable of causing a wide range of infections.1,2 Aminoglycosides are often used synergistically in combination with beta-lactam or glycopeptides to treat infections, especially for the treatment of complicated staphylococcal infection. 3 Concerningly, many methicillin-resistant S. aureus have acquired resistance to a wide range of antibiotics, including aminoglycosides. This has made the treatment of infections caused by this organism increasingly difficult. 4 Similarly, high-level aminoglycoside-resistant enterococci are becoming more prevalent and have caused significant problems for clinical anti-infection therapy. 5
Bacterial resistance to aminoglycosides is commonly mediated by three different mechanisms; enzymatic inactivation of the antibiotic molecule, decreased intracellular antibiotic accumulation, and the mutation of ribosomal protein or rRNA encoding genes. 6 In the clinical setting, production of aminoglycoside-modifying enzymes (AMEs) is one of the most frequently occurring mechanisms. AMEs catalyze modification at -OH or -NH2 groups of the 2-deoxystreptamine nucleus or the sugar moieties and can be acetyltransferases (AACs), nucleotidyltransferases (ANTs), or phosphotransferases (APHs). 7
The most commonly found gene coding for AMEs in Gram-positive bacteria is aac(6′)-Ie-aph(2″)-Ia, conferring resistance to a broad spectrum of aminoglycosides. Ant (4′)-Ia, aph (3′)-IIIa, and ant (6)-Ia genes that encode ANT(4′)-I, APH(3′)-III, and streptomycin modifying enzyme ANT(6)-I, respectively, are also important resistance genes.8–10 ANT(9)-Ia is characterized by conferring resistance to spectinomycin only.11,12 The gene encoding this enzyme was first cloned from Staphylococcus transposon TnS54. It was initially reported as being unique to S. aureus 12 and not observed in Enterococcus spp. 13 However, in a subsequent study, ant(9)-Ia was detected in clinical isolates of enterococci. 14 In addition, several new aminoglycoside resistance genes have been detected in enterococci, including aph(2″)-Ib, aph(2″)-Ic, aph(2″)-Id, and aph(2″)-Ie.14,15 In this study, we focus on the simultaneous detection of the most prevalent AME genes in both staphylococci and enterococci.
To implement appropriate infection control measures, public health laboratories perform surveillance of the precise types of antibiotic resistance genes present. Often, this genotyping depends on real-time PCR or single or multiplex end point PCR. These technologies have inherent limitations when applied to high-throughput detection. The Luminex xTAG assay is a multiplex assay that uses commercially available colored beads to perform biological assays similar to nucleic acid hybridization assays. The colored beads are 6.5 mm carboxylated, superparamagnetic polystyrene microspheres that are internally labeled with a spectrally distinct fluorescent dye. It provides a multiplexed bead array system capable of analyzing and reporting up to 150 different nucleic acid targets in a single reaction (www.luminexcorp.com). Each microsphere set is conjugated to antiTAG sequences, which can bind to complementary synthesized tags on specific primer sets. The reverse primers were biotin-labeled to combine with streptavidin-R-phycoerythrin (SAPE). If the target is present, these specific PCR products will be immobilized to the bead by hybridization between the tag and anti-tag sequences and the biotin bonds with SAPE. Because of the microscopic size and low density, the beads can be individually analyzed in a flow cytometry based detection system.
In this study, a novel multiplex Luminex xTAG assay was developed for the simultaneous detection of five prevalent aminoglycoside resistance genes in Gram-positive bacteria staphylococci and enterococci. The genes aac(6′)-Ie-aph(2″)-Ia, aph(3′)-IIIa, ant(4′)-Ia, ant(9)-Ia, and ant(6)-Ia were detected (Table 1). To validate the assay for use in routine settings, we applied the methods to 22 clinical bacterial isolates and compared the results to those obtained using published methods for AME gene detection using single or multiplex PCR.
Aminoglycoside-Modifying Enzyme Genes Detected in the xTAG-Multiplex PCR Assay
aac(6′)/aph(2″) is short for aac(6′)-Ie-aph(2″)-Ia.
AME, aminoglycoside-modifying enzyme.
Materials and Methods
Bacterial strains
All bacterial clinical strains used in this study are listed in Table 2. Twenty-two clinic bacterial isolates were isolated from patients of the Affiliated Hospital of Guangdong Medical University, Zhanjiang. They were all resistant to one or more aminoglycoside antibiotics (Table 2). Escherichia coli ATCC 25922, used as a negative control, was bought from the Guangdong Culture Collection Center. Five positive reference strains were identified by conventional PCR and stored in our laboratories (Supplementary Table S1). Bacterial cultures were grown overnight at 37°C on nutrient agar medium (Bio-Rad Laboratories). Nucleic acids were extracted from cell pellets with a TIANamp Bacterial DNA Kit (Tiangen Biotech, China) following the manufacturer's instructions. The concentration and purity of the DNA extracts were determined by measuring the A260/A280 optical density ratio with a NanoDrop 2000C spectrophotometer (Thermo Scientific).
xTAG Multiplex PCR Assay Screening Results for 22 Clinical Isolates
All isolates were from patients of the Affiliated Hospital of Guangdong Medical University, Zhanjiang.
+++, strong positive (MFI >5*cutoff); ++, positive (3*cutoff < MFI <5*cutoff); +, weak positive (cutoff < MFI <3*cutoff); −, negative (MFI < cutoff); +/−, weak positive for xTAG multiplex PCR assay but negative for agarose gel; −/+, negative for xTAG multiplex PCR assay but positive for agarose gel; Ak, amikacin; Gm, gentamicin; K, kanamycin; MFI, median fluorescent intensity; N, neomycin; Spcm, spectinomycin; Sm,streptomycin; Tob, tobramycin.
Primer design
PCR primers (Table 3) were designed using Primer Premier 5.0 software based on the sequence of the target genes uploaded from the GenBank database. The Primer-BLAST programa was used to analyze the specificity of the designed primers.
Oligonucleotide Primers Used for xTag Multiplex and Conventional PCR
The oligonucleotides underlined were used for standard PCR when recombinant plasmids were constructed.
The oligonucleotides in italics were used for conventional PCR.
Primers were tagged according to the instructions provided by the manufacturer. All primers were synthesized and then purified by high-performance liquid chromatography (Sangon Biotech, China).
Multiplex PCR amplification and xTAG assay
The pentaplex nucleic acid amplifications were performed using the QIAGEN Multiplex PCR Plus Kit (Qiagen). All reactions were performed in a 20 μL volume containing 10 μL Multiplex PCR Master Mix, 0.1 μg nucleic acid template, 250 nM of each PCR primer [except 300 nM for ant(9)-Ia], and ddH2O. Cycling conditions were: 95°C for 5 minutes, 35 cycles of 95°C for 30 seconds, 60°C for 90 seconds, and 72°C for 30 seconds, and 68°C for 8 minutes. Subsequently, 5 μL of the reaction mix was added to 75 μL of SAPE (1 mg/mL) (Thermo Fisher) and 20 μL of TAG beads (∼2500 beads). The hybridization was performed at 42°C for 25 minutes, under low light conditions.
Analysis was performed using a Luminex 200™ instrument (Luminex Corporation). The bead type was set to MagPlex, and the gate opening range was set as default with a plate heater temperature of 42°C. The median fluorescent intensity (MFI) values per target for each reaction were calculated using the Luminex xPONENT3.1 software supplied with the instrument. The MFI was calculated by the software after counting at least 100 beads for each bead set in a sample, as recommended by the user manual. 16 Negative controls containing all the hybridization components except target DNA were used in all experiments to define cutoff MFI values as the mean of the net MFI values of two negative PCR controls +3SD. 17 All samples were used in duplicate. When the net MFI values in both duplicates were above the cutoff, the sample was defined as positive.
To obtain well-characterized positive controls and determine the sensitivity of the xTAG multiplex PCR assay, a 220-bp fragment of aac-aph(2″)-Ia, a 146-bp fragment of aph(3′)-IIIa, a 119-bp fragment of ant(4′)-Ia, a 136-bp fragment of ant(9)-Ia, and a 114-bp fragment of ant(6)-Ia were amplified using standard PCR. All reactions were performed in a 20 μL volume containing 10 μL Premix Ex Taq HS (Takara, Japan), 0.1 μg nucleic acid template, 500 nM of each PCR primer, and ddH2O. Cycling conditions were as follows: 95°C for 5 minutes, 35 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 15 seconds, and 72°C for 8 minutes. These PCR products were then cloned into the pMD-19T vector (Takara). All of these cloned genes were sequenced by Sangon Biotech (China).
Conventional single or multiplex PCR
The presence of aac-aph(2″)-Ia, aph(3′)-IIIa, and ant(4′)-Ia was examined by multiplex PCR. 18 The reactions were conducted using the DNA polymerase provided in a Premix Ex Taq HS (Takara).The mixture consisted of 400 nM of each primer and 0.2 μg template DNA. The cycling parameters were as follows: 95°C for 3 minutes; 35 cycles of 94°C for 40 seconds, 55°C for 40 seconds, 72°C for 40 seconds, and a final extension at 72°C for 5 minutes. Conditions of two single PCRs were kept as described previously.8,14
To ensure the reproducibility of the results, all reactions were repeated at least thrice independently. PCR products were analyzed by electrophoresis at 100 V for 30 minutes in a 2% agarose gel stained with ethidium bromide.
Results
In this study, a pentaplex xTAG PCR assay was developed and compared to conventional PCR detection with 22 clinical bacterial isolates. In the xTAG multiplex PCR assay, aac(6′)-Ie-aph(2″)-Ia was the most frequently detected gene, found in 13/22 isolates (59.1%) (Table 2). The xTAG assay was in agreement with conventional PCR results in 95.5% of cases (Supplementary Fig. S1), with just a few minor discrepancies (Table 2). Three weak positive samples, including aac(6′)-Ie-aph(2″)-Ia of isolate 13, ant(4′)-Ia of isolate 16, and ant(9)-Ia of isolate 16, were missed by conventional PCR and agarose gel analysis. Ant(9)-Ia of isolate 20 was positive in agarose gel but negative in xTAG assay. The 838-bp PCR products were confusing in some clinic samples especially from isolate 17, which showed a product near the expected size. Subsequent nucleotide sequencing of the two PCR products showed that they were not consistent with the targets. The isolate tested negative in the multiplex Luminex xTAG assay (Supplementary Fig. S1). Our results showed that xTAG assay provided little higher detection rates and accuracy for AME genes than conventional PCR.
The sensitivity of the xTAG assay was evaluated using recombinant plasmids containing cloned target genes. All five templates were presented in each reaction mixture, and the limit of detection was determined to be 101 copies/μL for ant(6)-Ia and ant(4′)-Ia, 10 2 copies/μL for aac-aph(2″)-Ia and aph(3′)-IIIa, and 103 copies/μL for ant(9)-Ia (Fig. 1).
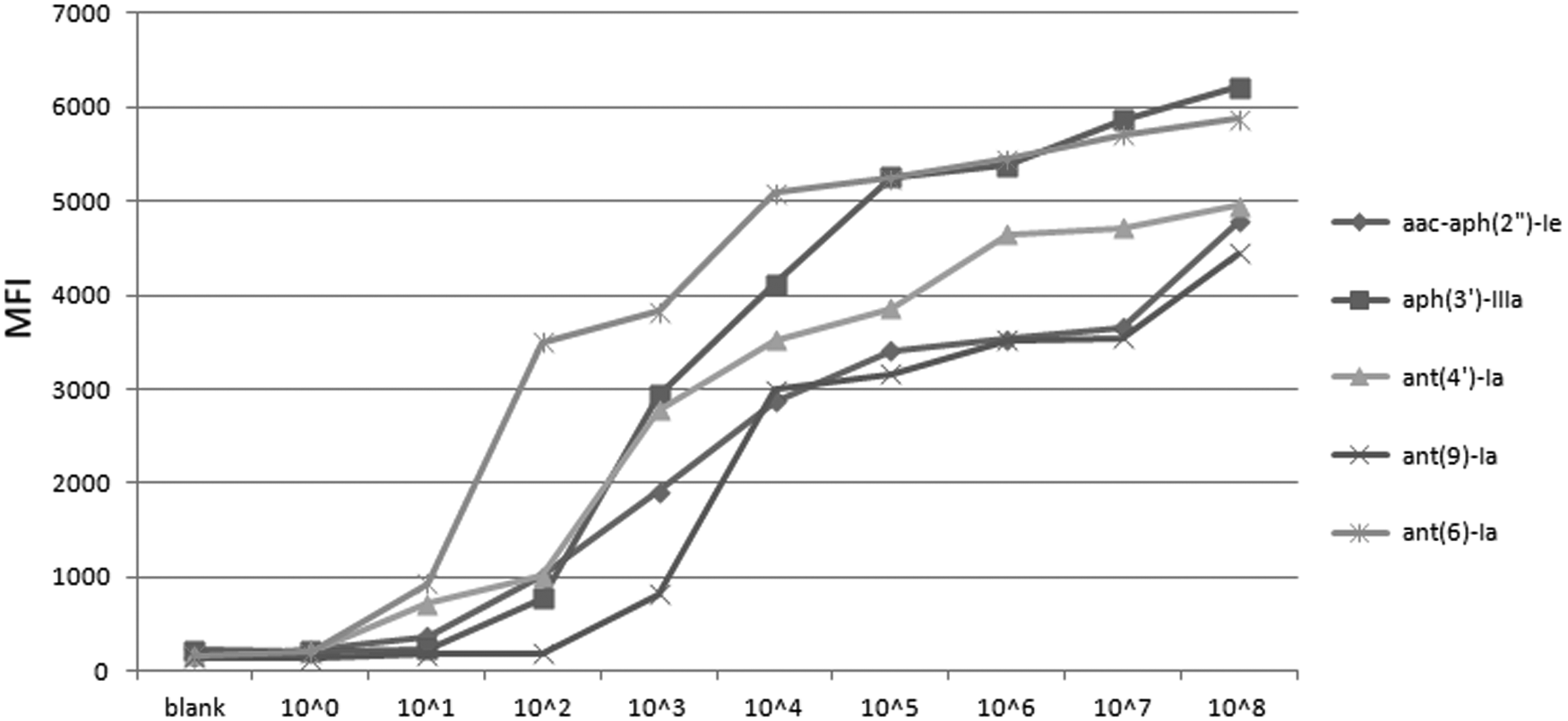
Sensitivity of the xTAG multiplex PCR assay. Serial dilutions of aac(6′)-Ie-aph(2″)-Ia, aph(3′)-IIIa, ant(4′)-Ia, ant(9)-Ia, and ant(6)-Ia recombinant plasmids. Blank: no template control.
The results obtained with conventional multiplex PCR assay were affected by different conditions, including the DNA extraction method, the concentration of primers, the Mg2+ concentration, and annealing temperature (data not shown). Also due to the intrinsic detection limits of agarose gel electrophoresis, some weak products were missed.
Discussion
In the database of resistance genes, hundreds of AME genes have been recorded.b For initial development of the new assay five of the most frequently found AME genes were selected for analysis. All of these genes were detected in Gram-positive bacteria enterococci14,19 and staphylococci. 20 The genes encode the main kinds of aminoglycoside resistances, including gentamicin, kanamycin, neomycin, lividomycin, paromomycin, butirosin, amikacin, isepamicin, dibekacin, streptomycin, spectinomycin, and ribostamycin (Table 1). In previous study, these five AME genes were found in plasmids, transposons, or chromosomes. Among them, aac-aph(2″)-Ia was found both in transposon and plasmid, ant(4′)-Ia was found in plasmid, and ant(9)-Ia and ant(6)-Ia were detected in transposons. 7 Total DNA from clinical isolates was used as template in this study to ensure that both plasmid and chromosomally encoded genes would be detected. In addition, we also tested for the presence of these genes in Pseudomonas aeruginosa, E. coli, and Klebsiella pneumoniae. As expected, no target genes were detected in these Gram-negative bacteria (Table 2). It is noteworthy that ant(9)-Ia was detected in five enterococci samples, reconfirming that ant(9)-Ia does occur in enterococci and is not only found in staphylococci. This indicates that the resistance genes are spread cross-boundary, and effective surveillance for prevalence of resistance genes needs to be established.
A previous study of the epidemiology of aminoglycoside resistance genes in enterococci developed a multiplex PCR for the detection of six aminoglycoside resistance genes aac(6′)-Ie-aph(2″)-Ia, aph(2″)-Ib, aph(2″)-Ic, aph(2″)-Id, aph(3′)-IIIa, and ant(4′)-Ia. PCR products ranged in size from 71 to 867 bp to distinguish these genes by gel electrophoresis. The multiplex PCR procedure was a convenient method for rapid detection of the presence of six aminoglycoside resistance genes in enterococci. 19 In this study, aph(2″)-Ib, aph(2″)-Ic, and aph(2″)-Id were not included in our assay as they have not been reported in staphylococci. It should be noted that the primer concentration used for each gene was as low as 5 pmol, and the sensitivity of multiplex PCR was not evaluated in the report. Due to the intrinsic limitations of traditional multiplex PCR assays, a negative result may not always signify the absence of a gene. Results were affected by the DNA extraction method, the concentration of the primers, the Mg2+ concentration, and annealing temperature.
Although the xTAG-multiplex PCR method also depends on nucleic acid amplification, for PCR product detection the multiplex Luminex xTAG assays rely on the “antiTAG” sequences conjugated to each microsphere rather than the PCR product size as determined by gel electrophoresis. The five PCR products were all designed to be around 200 bps in length to keep amplification and hybridization uniform for all the genes. The detection limits of the xTAG-multiplex PCR method were between 1 × 101 and 1 × 103 copies/μL. It was less desirable that there was a variation in the detection limits for each individual test within the multiplex assay. We assume that this is caused by differences in the amplification efficiencies of different primer pairs or possibly interference between primers since different primers. We have optimized the PCR condition to some extent but there may still be further scope to further improve and expand the capabilities of the assay. The sensitivity of ant(9)-Ia detection was lower than the other genes, and this may lead to some false negatives of weakly positive cases. However, when clinical samples were analyzed, the xTAG-multiplex PCR assay detected more AME genes than conventional PCR. In this study, the weakly positive cases in which there was poor amplification of targets may have resulted from low-quality nucleic acids or sequence variation in the primer regions. The xTAG-multiplex PCR assay is a new tool for the evaluation of AME gene presence in pathogenic bacteria. It costs less than $4.50 per assay and takes 4 hours to simultaneously detect five genes in bacterial clinical isolates, from DNA extraction to the final test result. It offers cost-effectiveness and good sensitivity as a high-throughput screening method of aminoglycoside resistance genes.
In summary, we developed a new multiplexed platform for simultaneous detection of five of the most common AME genes in clinical isolates of enterococci and staphylococci. The detection system can be easily implemented in any laboratory equipped with the appropriate devices. It is able to provide an easy-to-use method to determine what aminoglycoside might be efficacious in a clinical setting.
Footnotes
Acknowledgments
The study was supported by Science and Technology Research Program (2016A040403058) and (2017A030303025) from Department of Science and Technology, Guangzhou. This work was also funded by Science and Technology Planning Project of Guangdong province (2017B030314171).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.