
Abstract
In recent decades, Neisseria gonorrhoeae has developed resistance to several antimicrobial classes. Molecular epidemiology approaches are useful for detecting emerging, often resistant, gonococcal clones. In this study, 67 N. gonorrhoeae isolates from different anatomic sites, collected over 8 years in Italy, were analyzed by whole genome sequencing (WGS). WGS was performed using the Illumina NextSeq 500 platform. Phylogenetic analysis was based on core single nucleotide polymorphism (SNP) and core genome multilocus sequence typing (cgMLST). N. gonorrhoeae multi-antigen sequence typing (NG-MAST), MLST, and N. gonorrhoeae sequence typing for antimicrobial resistance (NG-STAR) were carried out in silico using WGS data. Antimicrobial susceptibility against a four-drug panel was evaluated using a gradient diffusion method. Overall, gonococci clustered in accordance with NG-MAST, MLST, NG-STAR, and antimicrobials susceptibility profiles, but not with the site of isolation, HIV status, and patient sexual orientation. Phylogenetic analysis identified nine clades: two of them were the predominant and including gonococci of G1407 and G2400 genogroups.
Introduction
The sexually transmitted pathogen Neisseria gonorrhoeae annually causes 78 million gonorrhea cases. 1 The European Centre for Disease Prevention and Control (ECDC) reported 75,349 cases in 27 European Union/European Economic Area (EU/EEA) countries 2 in the year 2016; of these cases 760 were notified by Italy. 2
The global spread of gonococcal antimicrobial resistance (GONO-AMR), including multidrug-resistant (MDR)3–5 and extensively drug-resistant (XDR) strains,6,7 represents a continuous challenge that may even lead to consider gonorrhea as an untreatable bacterial infection.
Treatment failure against extended-spectrum cephalosporins (ESCs) and azithromycin, the current combined treatment option, was also reported.8–15 Moreover, the pathogen can infect the urogenital tract, rectum, and pharynx through sexual contact, and extragenital infections may facilitate the acquisition or development of mutations associated to AMR.16,17
Because the surveillance of resistant gonococci at national and international levels is crucial for controlling gonorrhea, the ECDC implemented the European Gonococcal Antimicrobial Surveillance Programme (Euro-GASP), collecting data from 25 EU/EEA Member States including Italy. 4 In our country, data on GONO-AMR are reported through a network coordinated by Istituto Superiore di Sanità (ISS) hosting the National Reference Laboratory (NRL).
ESC resistance, cefixime, and ceftriaxone, together with azithromycin and ciprofloxacin resistance, involve several mutations in penA, mtrR, porB, ponA, gyrA, parC, and the 23S rRNA genes.17–20 N. gonorrhoeae multi-antigen sequence typing (NG-MAST) and multilocus sequence typing (MLST) have been broadly used to type N. gonorrheae.21,22 In accordance, a strong association between genogroup (G)1407 (including ST1901 by MLST) and isolates resistant to cefixime, ciprofloxacin, and/or characterized by azithromycin susceptibility closed to resistance values have already been described.10,21,23,24 Similarly, a significant association between G2400 and ciprofloxacin-resistant gonococci was identified. 25
Recently, the N. gonorrhoeae sequence typing for antimicrobial resistance (NG-STAR), based on sequencing of seven genes associated with AMR (penA, mtrR, porB, ponA, gyrA, parC and 23S rRNA), has been introduced for a comprehensive typing of resistant gonocoocci. 26
Advances in high-throughput whole genome sequencing (WGS) have increased the amount of available data for N. gonorrhoeae, favoring phylogenetic analysis by single nucleotide polymorphism (SNP) and the investigation of antibiotic-resistant target genes. 27
Hereby, WGS was used to determine the genomic characteristics, using core SNP and core genome MLST (cgMLST) analyses, of a subsample of 67 N. gonorrhoeae genomes from isolates collected from different anatomic sites within the NRL for GONO-AMR in Italy, between 2007 and 2014.
Materials and Methods
Isolates and antimicrobial susceptibility testing
A subsample of 67 gonococci collected in Italy, between 2007 and 2014, from 14 collaborating laboratories (clinics for sexually transmitted diseases, infectious diseases, gynecology, dermatology-venereology, combined service, and other laboratories) was selected. The isolates were from different anatomic sites: 17 from pharynx, 21 from rectum, and 24 from urethra. Five gonococci causing disseminated gonococcal infection were also included. For three patients, more than one isolate was collected.
Primary isolation, identification, and collection of gonococcal isolates, following standard microbiological procedures, were performed locally by the collaborating laboratories. The isolates were stored at −80°C in brain–heart infusion medium (Oxoid Ltd., Italy) containing 20% glycerol and sent to the NRL at ISS for further molecular investigations. Ethical approval was not required as clinical isolates were collected, processed, and stored as part of routine clinical care by the collaborating laboratories participating in the network. Patients' anonymous data were analyzed using EpiInfo software, version 3.3.2.
Antimicrobial susceptibility tests were performed after growth on Thayer–Martin medium (Oxoid Ltd.) with 1% IsoVitalex (Oxoid Ltd.) at 37°C in a 5% CO2 atmosphere, following the Euro-GASP guidelines. 28
Etest (bioMérieux, Sweden) and MIC Test Strip methods (Liofilchem Diagnostici, Italy) were carried out in agreement with the manufacturer's instructions to determine the antimicrobial susceptibility against four-drug panel (cefixime, ceftriaxone, ciprofloxacin, and azithromycin). The minimum inhibitory concentration (MIC) values were interpreted referring to EUCAST clinical breakpoint (version 8.1, 2018). 29 Isolates with MIC range value for cefixime of 0.094–0.125 mg/L were classified within the decreased susceptibility (DS) category 13 ; those with ciprofloxacin MIC ≥32 mg/L or azithromycin MIC ≥256 mg/L were defined as high-level resistant (HLR) gonococci. 30
The World Health Organization (WHO) N. gonorrhoeae G, K, M, O, and P reference strains were included in each assay. 30
WGS and assembly
Chromosomal DNA was extracted using the QIAamp DNA minikit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Multiplex libraries were prepared using the Illumina Nextera XT protocol. Paired-end, 150-bp indexed reads were generated on the Illumina NextSeq 500 platform (NextSeq500 Mid Output kit, 300 cycles) yielding an average of 1.8 M paired reads/sample and an average genome coverage of 260 × .
The raw sequence data quality was checked using FastQC. 31 High-quality bases (Q score >25) were kept and reads were trimmed using the software Sickle. 32 De novo assembly was carried out with the ABySS software version 1.5.2 (k parameter = 63). 33 Contigs longer than 500 bp were selected using an ad hoc script and kept for further analysis. The final assembly ranged from 276 to 687 (median = 396) contigs/sample covering ∼2.06 Mb of the N. gonorrhoeae genome.
Core SNP analysis
Phylogenetic trees were constructed using a core SNP alignment using the kSNP3 program. 34 Sixty-seven draft genomes obtained from clinical isolates plus 10 available on the Neisseria Sequence Typing Home Page* from international reference strains (WHO-F, WHO-G, WHO-K, WHO-L, WHO-M, WHO-N, WHO-O, WHO-P, ATCC 49226 and FA1090, except for ATCC 49226 genome, available on Public Health England) were aligned.
SNP discovery was based on k-mer analysis; therefore, no multiple sequence alignment was required. Moreover, kSNP3 removes k-mers within each genome that would result in allele conflicts, thus eliminating SNPs located in repetitive DNA regions. To identify the optimum k-mer value for this set of N. gonorrhoeae data, the program Kchooser was used before running kSNP3, and the resulting k-mer was 23. After the kSNP analysis, 7,778 core SNPs were identified. The core_SNP_matrix.fasta output file, containing only SNP loci located in the core genome, was used for further analysis. Maximum likelihood analysis, based on the Tamura-Nei model 35 was constructed in MEGA7 36 using as input the core SNP matrix output of kSNP3 and applying the bootstrap method with 500 bootstrap replications. Phylogenetic clades were assigned using PhyloPart with a percentile distance threshold value of 0.10. Evolutionary analysis was performed with MEGA736 using the maximum composite likelihood model. 37
cgMLST and whole genome MLST analyses
Genomes were analyzed and compared using the BIGSdb Genome Comparator implemented within the PubMLST.org database, 38 through the gene-by-gene analysis. A total of 1,668 loci between the 67 gonococci and the reference strains WHO-F, WHO-G, WHO-K, WHO-L, WHO-M, WHO-N, WHO-O, WHO-P, and FA1090 were compared as defined in the N. gonorrhoeae cgMLST scheme v.1.0. The reference strain ATCC 49226 was not included in this analysis because it was not deposited in the Neisseria PubMLST database.
Alleles at each locus were designated with an integer, identifying isolates with the same or different allelic profiles, and a distance matrix was generated based on the number of variable alleles resolving isolates into networks using the Neighbor-Net algorithm and generated by SplitsTree4 (version 4.13.1). 39
Incomplete loci were automatically removed from the distance matrix calculation for the Neighbor-Net graphs. 39
Supplemental material
The same cgMLST procedure was used for the whole genome (wg) MLST analysis based on 2,805 loci of the 67 genomes. A total of 533 loci, missing in all isolates, were excluded from the analysis together with the incomplete loci.
Molecular typing
WGS data were used for in silico NG-MAST, MLST, and NG-STAR typing.
The porB and tbpB alleles were assigned by the NG-MAST website. 22 Moreover, typing sequences were submitted to the Neisseria MLST website for sequence type (ST) determination.
Finally, the NG-STAR Canada website was used to obtain the penA, mtrR, porB1b, ponA, gyrA, parC, and 23S rRNA allele and the ST. The same website was used to submit new alleles and/or new allele combinations. The new alleles found in all the typing genes, except for gyrA and parC, were confirmed by Sanger method. To do this, primers and amplification parameters, as already described,22,40,41 were used.
Closely related STs were defined as belonging to the same genogroup, referring to published definition. 19 In particular, ST sharing one allele and showing >99% similarity in the other allele (≤5 bp difference for porB and ≤4 bp for tbpB) were included in the same genogroup. 21 Multiple sequence and amino acid alignments were obtained using CromasPro version 1.15 and Clustal Omega website.
Results
Patients data
Overall, 63 patients had at least one positive culture for N. gonorrhoeae; of them, 3 patients had more than 1 positive culture. Most isolates were from men (59/63; 93.6%), with a median age of 34.3 years, and 4 from women (4/63; 6.3%), with a median age of 32.5 years. Men who have sex with men (MSM) represented 69.3% (43/62) of the patients. For one patient the sexual orientation was not available. The majority of participants with gonococcal infections were Italians (56/63; 88.8%), and 6.6% (4/60) were HIV positive. For three patients the HIV status was not available.
Antimicrobial susceptibility
Table 1 gives the MIC range values for cefixime, azithromycin, ciprofloxacin, and ceftriaxone of gonococci by clades and year of isolation. For cefixime, the lowest value was 0.016 mg/L. Isolates with higher values clustered in clade IIIa, with the exception of one isolate with an MIC of 0.064 mg/L in clade Ic.
Minimum Inhibitory Concentration Range Values for Cefixime, Azithromycin, Ciprofloxacin, and Ceftriaxone by Clades and Year of Isolation
MIC, minimum inhibitory concentration.
In particular, in clade IIIa, except for four gonococci showing values of cefixime MIC lower (0.016 and 0.032 mg/L), all the isolates showed higher values: seven isolates with MIC range values of 0.047–0.064 mg/L, five showed a DS (0.094–0.125 mg/L), and three resistant (0.19 mg/L) profiles to cefixime. Those gonococci classified as cefixime DS were collected from the year 2009, and those that were cefixime resistant from 2011.
Azithromycin MIC values ranged from 0.006 to 0.5 mg/L, with the exception of one HLR isolate (>256 mg/L), clustering in clade IIb, collected in 2014. A total of 26 gonococci showed intermediate MIC values (0.38–0.5 mg/L) and were found in all but not in group I. Isolates with intermediate MIC values to azithromycin were collected during the study period.
The majority of gonococci were resistant to ciprofloxacin (n = 41; MIC >0.06 mg/L) and 24 were HLR (MIC >32 mg/L); they were dispersed in all clades, except clade IIIb and group I clades, and collected during all the study periods.
All gonococci were susceptible to ceftriaxone (MIC range values 0.002–0.064 mg/L); one isolate with higher value, collected in 2011, clustered in clade IIIa.
Core SNP combined with clinical, molecular, and antimicrobial susceptibility patterns
As given in Fig. 1, the maximum likelihood phylogenetic analysis grouped the 67 genomes into 9 clades (Ia, Ib, Ic, IIa, IIb, IIc, IIIa, IIIb, and IIIc) derived from three major groups (I, II, and III). Three most divergent genomes (ID1015, 2046, and 632) were out of the clades. The WHO international reference gonococcal genomes were interspersed out of the clades, except for WHO-K that clustered into clade IIIa and WHO-O into clade IIb. The number of amino acid substitutions per site from averaging over all sequence pairs between clades was evaluated, and all positions containing gaps and missing data were eliminated. There were 9,109 positions in the final dataset. Supplementary Table S1 estimates the evolutionary divergence over sequence pairs between clades obtained with core genome SNP analysis.
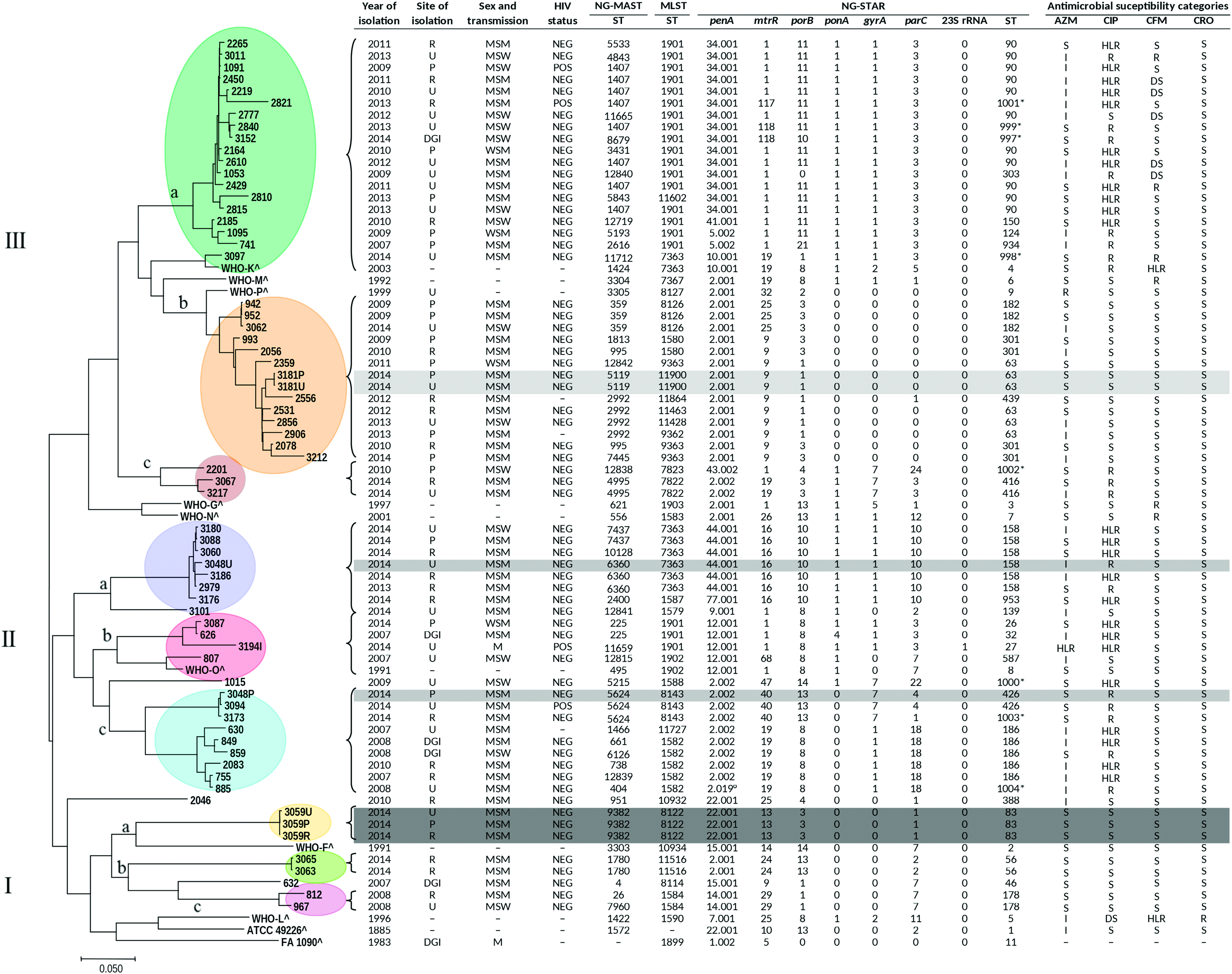
Maximum likelihood phylogenetic tree using core genome SNP of 67 genomes from Neisseria gonorrhoeae isolates, between 2007 and 2014, in Italy, and 10 genomes from N. gonorrhoeae reference strains. Year and site of isolation, sex and transmission, 4 HIV status, NG-MAST, MLST, and NG-STAR alleles and antimicrobial susceptibility categories were indicated. Roman numbers I, II and III refer to the group numbers; the different letters “a–c” refer to the clade's names. *New combination among the 7 NG-STAR alleles; °New allele; ^Information available exclusively for the reference strains: Ref.,26,30,54 and https://www.ebi.ac.uk/biosamples/; −, not available; DGI, disseminated gonococcal infection; DS, decreased susceptibility; HLR, high-level resistance; I, intermediate; MLST, multilocus sequence typing; MSM, men who have sex with men; NEG, negative; NG-MAST, N. gonorrhoeae multi-antigen sequence typing; NG-STAR, N. gonorrhoeae sequence typing for antimicrobial resistance; P, pharynx; POS, positive; R, in site of isolation, rectum; R, in antimicrobial susceptibility categories, resistant; S, susceptible; SNP, single nucleotide polymorphism; U, urethra; WSM, women who have sex with men. Color images are available online.
Within group I, the main clade was clade IIIa, including 19 isolates, collected half from heterosexual and half from MSM patients; the majority of them were HIV negative. As given in Fig. 1, the susceptibility profiles classified the isolates as DS (n = 5), resistant (n = 3), or susceptible (n = 11) to cefixime; intermediate (n = 10) or susceptible (n = 9) to azithromycin (n = 9) and resistant (n = 7), HLR (n = 11) or susceptible (n = 1) to ciprofloxacin.
Eleven isolates of clade IIIa (58%; 11/19) belonged to NG-MASTs 1407, 5533, 4843, and 3431 of genogroup G1407; ST1407 was the predominant (72.7%; 8/11). The 89.4% (17/19) of isolates belonging to clade IIIa were associated with ST1901 by MLST. penA allele 34.001, with the mosaic allele XXXIV, 26 in 79% (15/19) of the isolates was found together with the mtrR allele 1 (79%; 15/19), porB allele 11 (79%; 15/19). All isolates of clade IIIa had ponA allele 1, gyrA allele 1, parC allele 3, and wild-type 23S rRNA. NG-STAR ST-90 (n = 11) was identified in >50%. In clade IIIa, 4 new STs (ST-997, 998, 999, and 1001) were found and submitted as new combinations (NG-STAR Canada).
In the second group, clade IIa grouped eight gonococci, of which seven were collected from HIV-negative MSM. Two isolates were resistant, five HLR and one susceptible to ciprofloxacin. They showed an intermediate MIC value (n = 4) or susceptibility (n = 4) to azithromycin and susceptibility to cefixime and ceftriaxone (n = 8). Of the isolates, 87.5% (7/8) belonged to NG-MASTs 2400, 6360, 7437, and 10128 of genogroup G2400. Seventy-five percent (6/8) showed MLST 7363 and NG-STAR ST-158, comprising penA allele 44.001 (nonmosaic), mtrR allele 16 (-35 A deletion in the promoter), porB allele 10, ponA allele 1, gyrA allele 1, parC allele 10, and 23S rRNA wild type. The remaining two gonococci showed MLST 1587 associated with NG-STAR ST-953 and MLST 1579 with NG-STAR ST-139.
Clade IIb clustered four isolates of which one was collected from a male HIV-positive patient; the isolate resulted HLR for azithromycin (MIC >256 mg/L) and ciprofloxacin (MIC >32 mg/L) belonging to NG-MAST 11659, MLST 1901, and NG-STAR ST-27. The phenotypic resistant profile was confirmed by the presence of 23S rRNA allele 1 (comprising A2059G amino acid substitution in the four 23S rRNA alleles), gyrA allele 1 (S91F/D95G), and parC allele 3 (S87R).
Finally, within group I, three gonococci from the same MSM patient (HIV negative), collected from different sites of isolation (pharynx, urethra, and rectum), clustered in clade Ia. All isolates were susceptible to the antimicrobials tested. They showed identical molecular pattern: NG-MAST 9382, MLST 8122, and NG-STAR ST-83.
Differently, isolate ID3048U, collected from urethra, clustered in clade IIa, and isolate ID3048P, collected from the pharynx of the same patient, clustered in clade IIc, showing a different genomic profile (Fig. 1).
cgMLST and wgMLST analyses
Overall, 502 of the 1,668 core genome loci were included in the cgMLST analysis; the remaining 1,160 loci were incompletely assembled and 6 were missing in all the isolates. A total of 48 loci were identical.
As given in Fig. 2, the nine clades obtained by the core genome SNP analysis were confirmed by cgMLST. However, the genomes ID3097 and WHO-K were from clade IIIa.

Neighbor-Net phylogenetic network based on the comparison of 502 core genome loci (cgMLST) of 67 genomes from N. gonorrhoeae isolates and 9 N. gonorrhoeae reference strains. cgMLST, core genome multilocus sequence typing. Color images are available online.
A total of 737 loci were included in the wgMLST analysis of which 68 loci were identical in all isolates. As shown in Supplementary Fig. S1, the genomes clustered in the same clades, as obtained by cgMLST.
Discussion
Controlling N. gonorrhoeae infection is challenging, and the AMR of this pathogen is recognized as a public health issue internationally.3,5
N. gonorrhoeae has developed resistance against several antimicrobials that are considered in the current combined treatment option, comprising ESCs and azithromycin.8–13
Within the GONO-AMR routine surveillance, sequencing of AMR target genes along with NG-MAST typing5,21,22,25 is required for each resistant gonococcal isolate.
Nowadays, the role of WGS in improving the characterization of gonococci and quickly identifying emergent clusters of resistant or MDR isolates has been widely described and partially replaced by conventional single typing methods to monitor resistant gonococci.42–46
In Italy, in 2007–2008, a high percentage of azithromycin resistance 12 and cefixime resistance were described 47 as the main peculiarities of antimicrobial susceptibility patterns among gonococci isolated at that time. More recently, in 2009–2016, this profile has changed, 48 probably as a result of the implementation of recommended gonorrhea treatment guidelines, that include the use of dual therapy. 49 In particular, it is important to mention the decreased proportion of azithromycin-resistant gonococci observed until 201248; then, the resistance started to increase, 48 as also reported in other European countries. 4
The proportion of cefixime-resistant gonococci that was low from 2009 to 2011, remained quite stable until 2014, as documented in other European countries, 4 with a further decrease in 2016. 48
Of interest, despite the new treatment guidelines, the rate of ciprofloxacin-resistant gonococci maintains the same proportion during the years, representing, nowadays, ∼50% of all isolates.4,47
As previously reported,13,47 G1407 was predominant among gonococci collected in Italy. ECDC reported the spread of this international clone associated with resistant antimicrobial profile against cefixime and ciprofloxacin, but also a reduced susceptibility or resistance to azithromycin, 21 with an overall prevalence of 23.3%, from 2009 to 2010, in Southern and Eastern Europe. 21 After that, a lower proportion (14.8%) was reported in 2013, mostly in Southern Europe, Germany, and Scandinavian countries. 25 An additional emerging clone, G2400, mainly associated with ciprofloxacin resistance, was identified among the gonococci collected in Italy, as also reported in other EU countries. 25
The identification of predominant clades and of specific resistant clones, such as G1407, which were predominant between 2007 and 2014 is worthy of mention. Moreover, it is important to highlight how gonococci clustered regardless of the site of isolation and/or HIV status and/or patient sexual orientation. Likewise, similar to other studies, 50 we observed a high agreement between molecular traits and predictive antimicrobial phenotypes, as defined in vitro.
The results regarding the isolates of the main clade IIIa, comprising gonococci G1407 with DS or resistant to cefixime represent a relevant finding. It is important to consider that, in this clade, the lower cefixime MIC was 0.016 mg/L, but the majority of isolates showed a MIC ≥0.032 mg/L. Moreover, most of the isolates of G1407 were azithromycin intermediate (MIC range value of 0.38–0.5 mg/L) and ciprofloxacin resistant (MIC range value of 3–32 mg/L).
Similar to previous reports,10,13,23,24 those isolates of G1407 in clade IIIa collected in Italy over the time period, with mosaicism in penA gene (frequently penA-XXXIV), showed also the adenine deletion in mtrR promoter, the amino acid substitutions G120K and A121N in PorB, S91F and D95G in GyrA, and S87R in ParC.13,14,51,52
Half the G2400 isolates in clade IIa were intermediate to azithromycin; most of them were also resistant to ciprofloxacin and all were fully susceptible to cefixime. Furthermore, they did not show the mosaicism in penA or mutations in parC genes.
In this study, one isolate (belonging to NG-MAST 11659, MLST 1901, and NG-STAR ST-27) with HLR to azithromycin and ciprofloxacin was collected from an Italian male patient, 30 years old and HIV positive. As reported in the literature, HLR to azithromycin is associated with the amino acid substitution A2059G in the four 23S rRNA alleles.14,53
From the available data on the treatment, we know that the patients from whom clade IIIa gonococci was collected until 2012 had been treated with cefixime or ceftriaxone. The other patients with positive gonococcal isolation after that date were treated with ceftriaxone as monotherapy option. In only one case, in 2014, the treatment consisted of combined therapy (ceftriaxone plus azithromycin), as suggested in the recent international guidelines.
As previously discussed, 48 with the exception of the rare use of oral cefixime as monotherapy, in Italy the treatment of gonorrhea followed, for most cases, international guidelines. 49
Finally, genomic analysis may be useful to investigate also possible multiple gonorrhea infection. One of the examples is a case of gonorrhea in an MSM, HIV-negative patient, from whom two gonococci were collected: one from the pharynx and another from the urethra. The isolates, with different antimicrobial susceptibility profiles, clustered into two different clades, suggesting that these were two distinct isolates responsible for a concomitant N. gonorrhoeae infection.
As for other pathogens, WGS allows to explore the entire genome of N. gonorrhoeae and has proven to be an useful tool for molecularly predicting AMR profiles. WGS may also provide added value, rather than the routine typing schemes, within the surveillance system.
Footnotes
Acknowledgments
The Neisseria gonorrhoeae antimicrobial-resistant study group: I. Dal Conte: STI Clinic, Department of Infectious Diseases, Amedeo di Savoia Hospital, Turin, Italy; V. Ghisetti, S. Del Re, G. Gregori: Laboratory of Microbiology and Virology, Amedeo di Savoia Hospital, Torino, Italy, M.L. Stella: STI Clinic, Amedeo di Savoia Hospital, Turin, Italy; A. Di Carlo, A. Cristaudo, G. Prignano, M. Giuliani, A. Latini, A. De Santis, M.T. Gallo, M. Frasca: San Gallicano Dermatologic Institute, IRCCS, Rome Italy; M. Cusini, L. Corti, S. Ramoni, D. Fanoni: Foundation IRCCS Ca' Granda “Ospedale Maggiore Policlinico Milano,” Milan, Italy; M.A. De Francesco: “Dipartimento di Medicina Molecolare e Traslazionale, Sezione di Microbiologia,” University of Brescia, Italy; A. Matteelli, M. Gulletta: Clinic of Infectious and Tropical Diseases, University of Brescia, Italy; MP. Landini, A. D'Antuono, C. Vocale: Unit of Clinical Microbiology, CRREM Laboratory, St. Orsola-Malpighi, University Hospital, Bologna, Italy; R. Antonetti, A. Di Taranto, R. De Nittis: Department of Clinical Pathology, “Azienda Ospedaliero-Universitaria OORR,” Foggia, Italy; M. Gaino, P. Ober, R. Predazzer: Microbiology and Virology Laboratory, Santa Chiara Hospital, Trento, Italy; F. Urbani: U.O. Dermatology, Santa Chiara Hospital, Trento, Italy; M. Busetti, M.T. Bortolin: Microbiology Unit, University Hospital ASUITs, Trieste, Italy; S. Del Monte: STI Clinic of San Lazzaro Dermatological Hospital, “A.O. Città della Salute e della Scienza di Torino,” Turin, Italy; A.M. Barbui, S. Brossa: Microbiology and Virology Laboratory, Molinette Hospital, Turin, Italy; E. Stroppiana: Dermatologic Clinic, “A.O.U. Città della Salute e della Scienza,” Turin, Italy; A. Mencacci, L. Levorato: Microbiology Section, Department of Experimental Medicine, University of Perugia, Italy; M. A. Latino, E. Gaido: Department of “Medicina di Laboratorio,” “P. O. Sant'Anna, Città della Salute e della Scienza di Torino,” Turin, Italy; CL. Bonanno, M. C. Cava: Microbiology and Virology Laboratory, Sandro Pertini Hospital, Rome, Italy; F. Poletti: Department of Infectious Diseases, “Ospedali Castelli ASL VCO,” Verbania, Italy; M. Rotondi, M. Minuti: Marilab s.r.l., Ostia, Italy; IC Casonato, M. Ferlini: Clinical Patology/Microbiology Laboratory, “A. O. Mauriziano di Torino,” Turin, Italy; E. Pagani, R. Aschbacher, P. Innocenti: Microbiology and Virology Laboratory, Azienda Sanitaria dell'Alto Adige, Bolzano, Italy.
This publication made use of the Neisseria Multilocus Sequence Typing website developed by Keith Jolley and hosted at the University of Oxford. 38 The development of this site has been funded by the Wellcome Trust and European Union.
This work was supported by the Ministry of Health-CCM Project 2014 “Sorveglianza di laboratorio della farmaco resistenza di Neisseria gonorrhoeae come malattia emergente” and Project 2016 “Attività di interesse comune finalizzate alla continuazione e alla sistemazione delle sorveglianze sanitarie.”
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.