
Abstract
Increased use of colistin in both human and veterinary medicine has led to the emergence of plasmid-mediated colistin resistance (mcr genes). In this study, we report the development of a real-time PCR assay using TaqMan probe-based chemistry for detection of mcr genes from bacterial isolates. Positive control isolates harboring mcr-1 and mcr-2 yielded exponential amplification curves with the assay, and the amplification efficiency was 98% and 96% for mcr-1 and mcr-2, respectively. Each target gene could be reproducibly detected from a sample containing 103 cfu/mL of mcr-harboring bacteria, and there was no cross-reactivity with DNA extracted from several multidrug-resistant bacteria harboring other resistance genes, but lacking mcr genes. Both sensitivity and specificity of the mcr real-time PCR assay were 100% in a method validation performed with a set of 25 previously well-characterized bacterial isolates containing mcr-positive and -negative bacteria. This newly developed assay is a rapid and sensitive tool for detecting emerging mcr genes in cultured bacterial isolates. The assay was successfully validated according to quality standards of the Clinical Laboratory Improvement Amendments (CLIA).
Introduction
The emergence of carbapenem-resistant Enterobacteriaceae has led to increased use of polymyxins as a treatment option in both veterinary and human medicine worldwide. 1 Colistin confers broad-spectrum activity against Gram-negative bacteria and is effective against antibiotic-resistant bacteria causing infections that are otherwise difficult to treat. Colistin resistance was previously reported as chromosomally mediated and thus nontransferable. 1 However, in recent years, a plasmid-borne colistin resistance gene, mcr, has been identified. 2
First reported in Escherichia coli isolates from China in 2015, the mcr-1 gene was subsequently identified in E. coli and other Enterobacteriaceae from human, animal, foodborne, and environmental sources globally.2–4 An MCR-producing organism was first reported in the United States in 2016 4 and has since then been identified in multiple isolates, including E. coli, Klebsiella pneumoniae, and non-Typhi Salmonella enterica.5–7 Soon after the discovery of mcr-1, other mcr variants were detected. The mcr-1.2 variant was identified in K. pneumoniae from Italy in 2016, 7 and mcr-2 was identified in E. coli from Belgium in 2016. 8 To date, nine variants of mcr-1 (mcr-1.2–1.9) have been reported to the National Center for Biotechnology Information (NCBI),9–11 and additional mcr alleles, mcr-3, mcr-4, mcr-5, mcr-6, and mcr-7, have also emerged.12–16 Given that colistin is a last-resort treatment option for multidrug-resistant infections, the emergence and rapid dissemination of multiple mcr genes are causes for international concern. To stem the spread of mcr genes, rapid detection is crucial. In this study, we describe the development and validation of a multiplex real-time PCR assay that can be implemented to screen isolates for different mcr variants.
Materials and Methods
Bacterial isolates
Positive control isolates harboring mcr-1 (AR-0346) and mcr-2 (AR-0538) were obtained through the CDC and FDA Antimicrobial Resistance Isolate Bank * and were used to develop the assay. An additional 23 bacterial isolates (Enterobacteriaceae, Pseudomonas aeruginosa, and Acinetobacter baumannii) collected through surveillance and reference activities at the U.S. Centers for Disease Control and Prevention (CDC) were used to further validate the assay. For each isolate, antimicrobial susceptibility to colistin was confirmed by reference broth microdilution performed according to guidelines established by the Clinical and Laboratory Standards Institute (CLSI). 17 All bacterial isolates were grown overnight on nonselective agar, 5% sheep's blood tryptic soy agar (Fisher Scientific, Hampton, NH), at 35°C. DNA was extracted using either the Maxwell MDx™ (Promega, Madison, WI) or the thermal/NaOH method, as previously described by Conrad et al. 18 Extracted DNA was stored at −20°C until tested.
Real-time PCR assay
Oligonucleotide primers and TaqMan probes were designed according to published sequences (GenBank accession numbers NG_0504417 and NG_051171) to amplify a region of 106 and 100bp of mcr-1 and mcr-2, respectively (Table 1). The specificity of primers and probes was verified in silico by BLASTN analysis against the NCBI database. Universal 16S rRNA primers and probe were included as endogenous controls. 19 The mcr-1 and mcr-2 probes were labeled with FAM and HEX, respectively, whereas the 16S rRNA probe was labeled with CY5.
Primers and Probes Used for Detection of mcr-1 and mcr-2
[+A], locked nucleic acid.
[R], degenerate base (Purine A or G).
PCRs were processed on an ABI 7500 Fast system (Applied Biosystems, Foster City, CA) and contained 10 μL of 1 × KAPA Probe Fast qPCR Master Mix (KAPA Biosystems, Wilmington, MA), 5 μL of primer and probe mix, 3 μL of sterile water, and 2 μL DNA template to a total volume of 20 μL. End working concentration of primers was 500 nM for each of the mcr-1 and mcr-2 primers, 125 nM for each 16S primer, and 250 nM for each probe. Thermal cycling was performed using the following conditions: 3 min at 95°C and 35 cycles of 3 sec at 95°C and 30 sec at 60°C. A sample was considered positive if it displayed a cycle threshold (Ct) value of 10–30. Isolates containing mcr-1 and mcr-2 were used as positive controls in the assay. E. coli ATCC 25922 was used as a negative control and each run included a non-template control.
Assay evaluation
To determine amplification efficiency of the mcr-1 and mcr-2 targets, a tenfold serial dilution of each starting genomic DNA template was prepared and analyzed with the assay. Each dilution was run in triplicate. The obtained Ct values were plotted against the dilution factor and amplification efficiency was estimated from the slope of the regression line.
To further evaluate the assay, mcr-1-positive and mcr-2-positive isolates were extracted and combined in the same reaction to rule out cross-reactivity or competition between primers and probes.
In addition, suspensions of 0.5 McFarland (10 8 cfu/mL) were established for the mcr-1 and mcr-2-positive control isolates. Tenfold serial dilutions were performed on each suspension and plated to obtain a viable colony count. Genomic DNA was extracted from 100 μL of each dilution using a Maxwell MDx™ system (Promega, Madison, WI), and the multiplexed real-time PCR assay was performed with 2 μL of each indicated dilution. Each dilution was run in triplicate.
Assay validation
The multiplexed mcr assay was validated according to standards of Clinical Laboratory Improvement Amendments (CLIA) using a set of 25 previously well-characterized isolates containing both mcr-positive and -negative bacteria (Table 2). Assay reproducibility and accuracy were evaluated by having a subset of the 25 isolates analyzed in three separate runs by three different laboratorians.
Bacterial Isolates Used in mcr Assay Development and Validation
mcr-1+ indicates positive for MCR-1 primer.
mcr-2+ indicates positive for MCR-2 primer/probe set.
Ct, Cycle Threshold; MIC, Minimum Inhibitory Concentration; Std Dev, Standard Deviation.
Assay application
The assay was further used to retrospectively screen 290 Enterobacteriaceae surveillance isolates that were collected over multiple years (2013–2015) and displayed a colistin minimum inhibitory concentration (MIC) of ≥2 μg/mL.
Results
The two E. coli mcr-1 and mcr-2-positive control isolates yielded exponential amplification curves in the assay when run in monoplex and multiplex reactions. Overall, a mean Ct of 16.3 ± 0.6 and 17.9 ± 0.5 was obtained for mcr-1 and mcr-2, respectively (data not shown). By performing a tenfold serial dilution of the mcr-1 and mcr-2 starting DNA templates, Ct values for each dilution were determined to establish the PCR amplification efficiency for mcr-1 (Fig. 1a) and mcr-2 (Fig. 1b). By plotting Ct values against the dilution factor, mean amplification efficiency was determined to be 98% for mcr-1 (Fig. 1c) and 96% for mcr-2 (Fig. 1d).
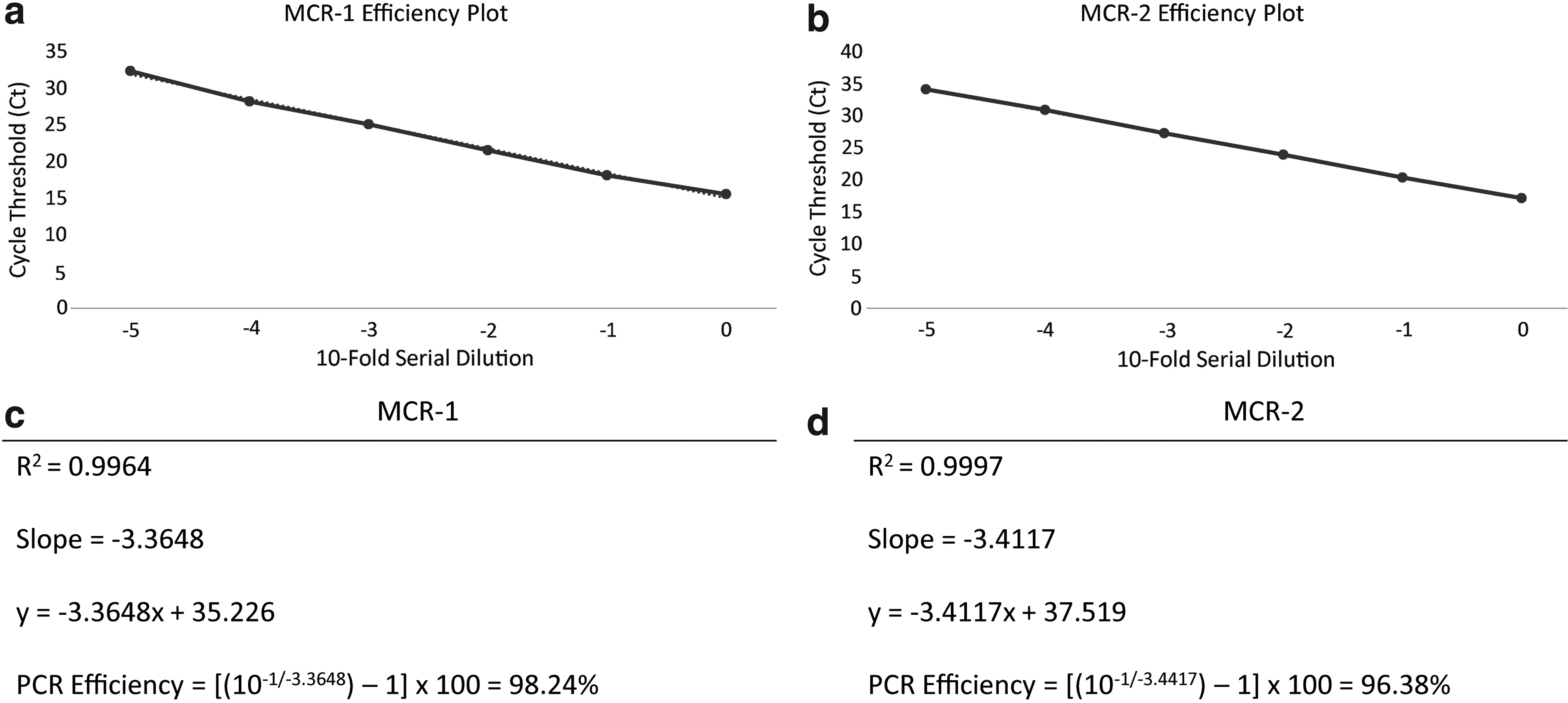
Efficiency plot of mcr-1
The assay was further challenged by combining the DNA of mcr-1 and mcr-2-positive isolates into the same reaction. Both targets were successfully detected with Ct values of 20.1 and 19.1, respectively.
Tenfold serial dilutions of 0.5 McFarland suspensions for both mcr-1 and mcr-2-harboring isolates were detected by real-time PCR over repeated runs (Tables 3 and 4). The real-time PCR assay could detect mcr-1 and mcr-2 from dilutions containing an average of 8.5 × 10 3 and 7.7 × 10 3 cfu/mL, respectively.
Ct Values for Samples Obtained from Tenfold Serial Dilutions of a 0.5 McFarland Suspension of the mcr-1-Positive Control Isolate (AR-0346)
For each 0.5 McFarland suspension, 100 μL was used to extract and elute DNA into 60 μL. Two microliters of eluted DNA was added to each PCR to determine Ct values.
TNTC, too numerous to count.
Ct Values for Samples Obtained from Tenfold Serial Dilutions of a 0.5 McFarland Suspension of the mcr-2-Positive Control Isolate (AR-0538)
For each 0.5 McFarland suspension, 100 μL was used to extract and elute DNA into 60 μL. Two microliters of eluted DNA was added to each PCR to determine Ct values.
To further validate the sensitivity and specificity of the assay, it was applied to a larger set of 25 well-characterized isolates, including six mcr-positive isolates (four mcr-1, one mcr-1.2, and one mcr-2). All mcr-positive isolates were successfully detected using the assay and the specificity was determined to be 100% as no false positive results occurred among the 19 mcr-negative isolates. Running a subset of isolates across three separate runs by three different laboratorians yielded consistent results, demonstrating that the assay was reproducible and accurate (Table 2).
No mcr-positive isolates were identified through additional screening of 290 Enterobacteriaceae isolates displaying a colistin MIC of ≥2 μg/mL (data not shown).
Discussion
Laboratory detection of colistin-resistant organisms is a challenge given the lack of phenotypic methods that will reliably detect these organisms. Disk diffusion does not work because of poor diffusion of the large colistin molecule. 20 In addition, gradient strips are not recommended since they have been shown to underestimate MIC values by one or more twofold dilutions relative to broth microdilution.21,22 To date, broth microdilution represents the only recommended method for colistin susceptibility testing. 22 However, colistin susceptibility testing is not available on commercial AST instruments. The lack of commercially available U.S. Food and Drug Administration (FDA)-cleared or -approved colistin susceptibility tests, along with the small number of clinical laboratories having the capacity to perform colistin susceptibility testing using broth microdilution, emphasizes the need for molecular tools to detect colistin-resistant organisms. 23
The newly developed assay described in this article represents a rapid tool for accurate, sensitive, and specific detection of mcr-1, mcr-1.2, and mcr-2 genes. The assay is highly efficient and reproducible with an efficiency of 98% and 96% for mcr-1 and mcr-2, respectively. While other real-time PCR assays have been developed to detect mcr-1,24,25 few multiplex assays have been developed to detect both mcr-1 and mcr-2. 26 In addition, by combining the DNA template of the mcr-1 and mcr-2-positive controls, we confirm that the present assay can detect both targets in the same reaction.
Implementation of the current assay would facilitate early and accurate detection of isolates carrying mcr genes. These genes are particularly relevant as they are plasmid-borne and can transfer within and between bacterial species. Containment of MCR-producing organisms is important to curb the spread of not only colistin resistance but also other antibiotic resistance genes as mcr-harboring bacteria often exhibit co-resistance to other antimicrobial agents. Of note, mcr-positive isolates harboring extended-spectrum β-lactamase-encoding genes have been reported from Europe, Asia, and South America. 27 Other reports of co-occurrence of blaCTX-M and mcr genes residing on the same plasmid have been reported.28–30 Finally, multiple reports have found mcr genes in carbapenemase-producing organisms,31–33 and others have confirmed the coproduction of MCR and a carbapenemase from a single plasmid.2,34
Multiple mcr-1 variants have been reported to NCBI [most recently mcr-1.7 (GenBank accession number ARJ33985.1), mcr-1.8 (GenBank accession number AQY61516.1), and mcr-1.9 (GenBank accession number ARA74236.1)]. Although other mcr-1 variants were not available for our study, in silico analysis confirmed that sequence variation is outside of the mcr-1 primer and probe target regions used in our assay. Therefore, we predict that the primers and probes described should detect all currently reported mcr-1 variants, although further studies will be required to confirm this.
To date, other mcr alleles have been reported, including mcr-3, mcr-4, mcr-5, mcr-6, and mcr-7.12–16 The mcr-3, mcr-4, and mcr-5 genes display between 71% and 76% sequence homology to mcr-1 and mcr-2.12–14 Based on in silico analysis, the primers and probes described in this article would not amplify mcr-3, mcr-4, or mcr-5; however, real-time PCR assays targeting these new alleles are currently under development in our laboratory.
In summary, we developed a sensitive and specific real-time PCR assay to detect mcr-1, mcr-1.2, and mcr-2 genes and successfully validated it to meet CLIA standards. This method will be a valuable tool for epidemiological studies assessing the prevalence of mcr-harboring bacteria as well as for rapid public health response to halt transmission of mobile colistin resistance. To support CDC's efforts in preventing antimicrobial-resistant infections in health care settings, the assay was shared with 56 U.S. state and local public health laboratories participating in CDC's Antibiotic Resistance Laboratory Network. To further facilitate early detection of mcr-containing bacteria, we encourage clinical laboratories with the capacity to perform reference colistin susceptibility testing to implement this assay to screen isolates displaying elevated MICs of colistin.
Footnotes
Acknowledgments
The authors would like to thank Dr. Anette Hammerum and Dr. Robert Skov for providing the mcr-1 isolate, Prof. Di Pilato for providing the mcr-1.2 isolate, and Prof. Xavier for providing the mcr-2 isolate.
Disclaimer
The findings and conclusions in this report do not necessarily represent the views of the Centers for Disease Control and Prevention.
Disclosure Statement
No competing financial interests exist.