
Abstract
Appropriate empiric therapy reduces mortality and morbidity associated with serious Gram-negative infections. β-lactams (BLs) owing to their safety, efficacy, and coverage spectrum are the most preferred agents for empiric use. Inappropriate use of older penicillins and cephalosporins led to selection and spread of resistant clones. As a result, these valuable agents have lost their reliability compelling clinicians to often use erstwhile last-line therapies such as carbapenems. Excessive carbapenems use imposed collateral damage by selecting difficult-to-treat carbapenem-resistant organisms. Lack of empiric therapeutic options amenable for use in infections caused by contemporary pathogens was realized by the pharmaceutical industry leading to intensive efforts in discovering novel antibiotics. These efforts led to the approval of newer β-lactams and β-lactamase inhibitor (BL-BLI) combination. This review elaborates the past trends in empirical use of BLs and ensuing patterns of resistance emergence in Gram-negatives. Furthermore, a critical appraisal of newer BL-BLIs has been presented to identify the appropriate clinical situations for their use to ensure clinical efficacy coupled with minimal resistance selection. These learning have been derived from past trends of clinical usage of older empiric therapies so that the therapeutic utility of newer agents is preserved for long in light of dwindling global antibiotics pipeline.
Introduction
Gram-negative bacterial pathogens are at the forefront of antibiotic resistance and often manifest multidrug resistance (MDR) in several clinically significant organisms. Treatment of infections caused by MDR Gram-negative isolates is often associated with poor or uncertain clinical outcome characterized by increased morbidity and mortality, extended length of hospital stay, eventually leading to higher health care cost. 1 To make the matter worse, in the past 20 years there has been a serious decline in discovery and development of MDR active Gram-negative antibiotics. It has been estimated that by 2050, if newer antimicrobial drug options are not made available then widespread resistance in bacterial pathogens would be responsible for ∼300 million deaths and drain in the world's economic growth (gross domestic product) to the tune of $100 trillion. 2
Empiric therapy in hospital settings for the treatment of range of infections caused by Gram-negative pathogens such as urinary tract infections (UTIs), intra-abdominal infections (IAIs), and hospital-acquired bacterial pneumonia or ventilator-associated bacterial pneumonia (HABP/VABP) is almost a norm.3,4 Choice of appropriate empiric therapy in critically ill patients is challenging as, at the initiation of therapy, the information on identity of infecting pathogen and its antibiotic susceptibility profile is not available. As a result, clinicians choose therapeutic options based on drug's extent of coverage of pathogen/resistance mechanisms and its potential effectiveness in light of hospital/regional level epidemiology. In such a scenario, it is desirable that a likely empiric therapy is bestowed with the features of broad-spectrum coverage of pathogen/resistance mechanisms to overcome varied patterns of local pathogen susceptibilities. Other features such as optimal pharmacokinetic/pharmacodynamic (PK/PD) at the site of infection as well as comforting safety profile ensure favorable clinical outcome. Additional factors such as ease of drug administration and dose adjustments (for special population) as well as lack of drug–drug interaction are also desirable attributes in an empiric setting. It has been widely recognized that a delay in instituting appropriate empiric therapy results in increased mortality, hospital stay, and cost of therapy. 5 With the availability of antibiotics and combination therapies with apparently overlapping pathogen coverage profile, it has always been challenging to make a right choice of an empiric therapy for a given clinical condition. Selection of a right empiric candidate is required for a favorable clinical outcome and minimal prospects of on-therapy resistance development.
Historically, the empiric management of Gram-negative bacterial infections involved the use of aminoglycosides, fluoroquinolones, and β-lactams (BLs) or β-lactam and β-lactamase inhibitor (BL-BLI) combinations. Among them, BLs are the most preferred agents in view of their excellent tolerability, broad-spectrum coverage, and consistent clinical efficacy for the treatment of range of Gram-negative infections. Moreover, several BL drugs are available in both oral and parenteral dosage forms facilitating their use in hospital as well as in community setting or as step-down therapies. This review would focus on trends in past clinical usage of BLs, their microbiological properties and PK/PD features and would also attempt to identify the possible reasons behind their erosion of efficacy due to resistance development. Later sections attempt to identify the appropriate clinical settings wherein the newer empiric agents (based on their microbiological/pharmacological features) could be deployed thereby possibly helping preserve their effectiveness for future.
Cephalosporins
In early years during 1940–1960, BL agents (mostly penicillins) were often deployed for the management of Gram-positive infections, which was posing higher degree of therapeutic challenge as compared with Gram-negative infections. During follow-on years, the rise of Gram-negative infections triggered the usage of cephalosporins, particularly those belonging to second and third generation as they were better optimized for Gram-negative infections. First-generation cephalosporins such as cephalothin, cephradine, and cefazolin were primarily active against Gram-positive pathogens and exhibited poor activity as well as suboptimal PK/PD with regard to Gram-negative pathogens. 6 However, due to limited number of therapeutic options available then, such agents were routinely used in hospital settings involving pre- and post-operative surgeries such as cholecystectomy, vaginal hysterectomy, and caesarean section, thereby exerting significant resistance selection pressure. 7 The problem was further aggravated by the use of first-generation oral cephalosporins such as cefadroxil and cephalexin in the treatment of Gram-negative infections even though they possessed features commensurate for the treatment of Gram-positive infections such as community-acquired bacterial pneumonia. This triggered emergence of narrow-spectrum cephalosporinases such as SHV-1 and TEM-1 (also called as older spectrum β-lactamases; Fig. 1 shows the observed relationship between BLs usage and their impact on the evolution of various β-lactamases).
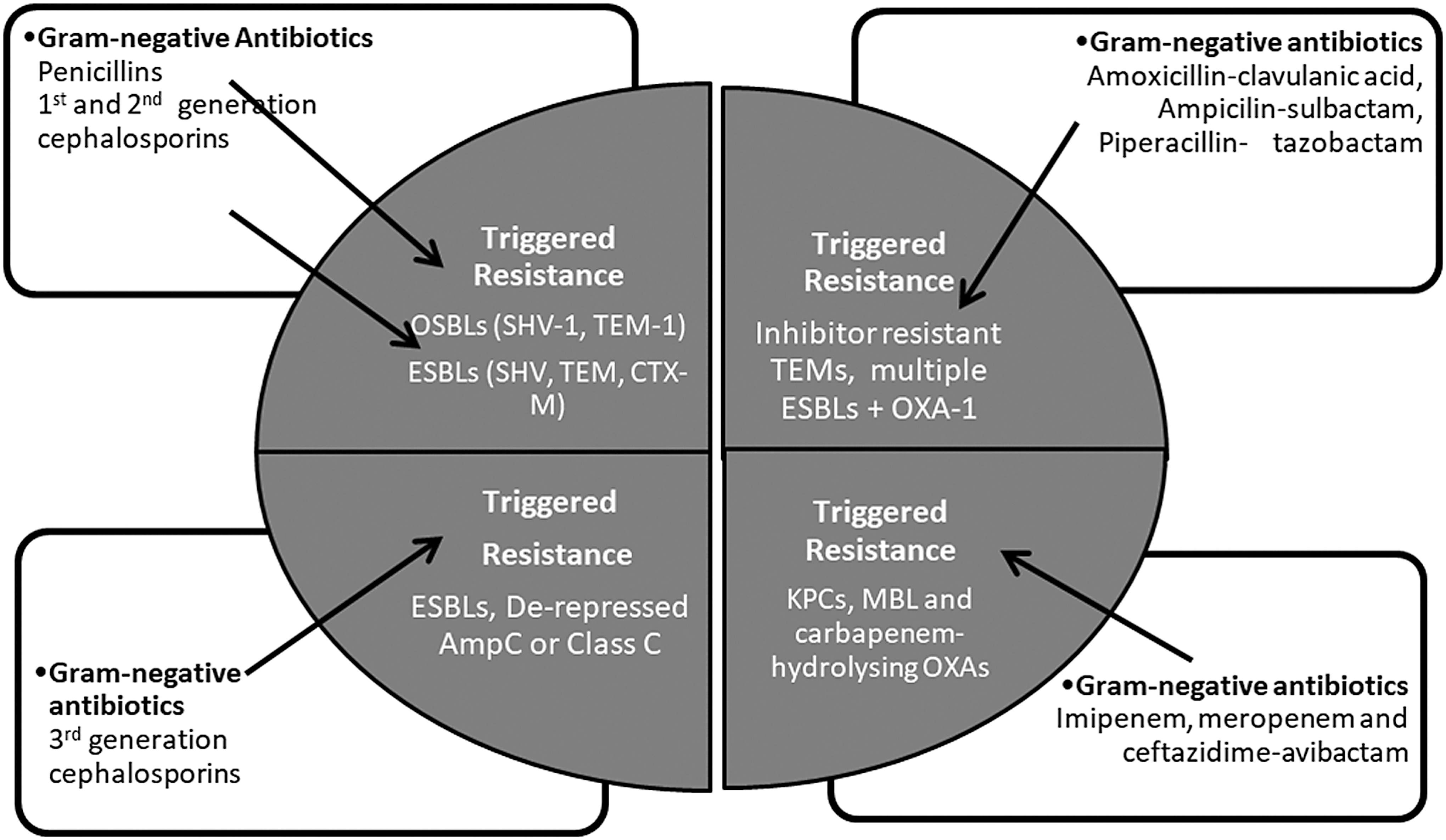
Schematic diagram depicting observed relationship between use of β-lactam antibiotics and triggered evolution of newer β-lactamases in Enterobacterales. ESBLs, extended spectrum β-lactamases; KPC, Klebsiella pneumoniae carbapenemase; MBL, metallo-β-lactamase; OSBLs, older spectrum β-lactamases.
Among second-generation cephalosporins, cefotetan, cefmetazole, and cefoxitin possessed moderate activity against Gram-negatives. 6 The PK properties of these second-generation cephalosporins are similar to that of first-generation cephalosporins and did not offer a significant therapeutic advantage against Gram-negative pathogens.6,8 Being active against anaerobic pathogens, excessive use of cefoxitin and cefotetan for the treatment of intra-abdominal and pelvic infections caused by Gram-negative anaerobic bacteria contributed in resistance development as such infections are invariably polymicrobial involving aerobic Gram-negative pathogens as well. 9
Third-generation cephalosporins, for the first time, provided well-optimized therapeutic options for the treatment of Gram-negative infections due to their potency and optimal PK properties. Ceftizoxime, cefotaxime, cefoperazone, ceftazidime, and ceftriaxone display broad-spectrum activity with improved activity against Gram-negatives particularly for ceftizoxime, cefoperazone, and ceftazidime. In addition, ceftazidime and cefoperazone are relatively stable to then prevailing narrow-spectrum cephalosporinases.10–12 Notably, ceftazidime offered additional advantage of enhanced activity against Pseudomonas aeruginosa.11,12 Even the PK properties (Cmax, half-life, and protein binding) of these drugs, especially for ceftazidime and cefotaxime, score over earlier cephalosporins.13,14
Among third-generation cephalosporin, ceftriaxone displays longer half-life of 5.8–8.7 hours (as a result of high serum protein binding of 85–95%), offering the PK features commensurate to once-a-day dosing. 15 However, the availability of low unbound drug concentrations of ceftriaxone through once-a-day dosing is adequate only for the treatment of infections caused by Gram-positive pathogens for which ceftriaxone displays high and consistent potency. 16 However, ceftriaxone was widely used in treatment of infections caused by Gram-negative pathogens resulting into several reports attributing the rise of extended spectrum β-lactamases (ESBLs) and AmpC β-lactamases to the irrational use of ceftriaxone (Fig. 1).17,18
In the hindsight, it seems that limiting the use of ceftriaxone and cefotaxime for community respiratory infections and that of ceftazidime for Gram-negative pneumonia would have significantly controlled resistance development. Third-generation cephalosporins further enhanced the selection pressure against inducible and derepressed AmpC β-lactamases encountered in Enterobacter spp. and Citrobacter spp., leading to emergence of derepressed mutants of AmpC (Fig. 1). 18 For instance, between 1983 and 1994, prevalence of derepressed AmpC in Enterobacter cloacae increased from 14.3% to 23.6% and in P. aeruginosa increased from 5% to 9% during the same period. 19 Even in the present times, the repercussions of spread of derepressed AmpC-based resistance mechanism are evident in the form of carbapenem resistance for strains co-harboring impermeability as another resistance mechanism.
Fourth-generation cephalosporins represented by cefepime and cefpirome are characterized by quaternary ammonium salt, which provides zwitterionic nature to these cephalosporins thereby imparting improved permeation features in Gram-negatives.20,21 Both the antibiotics provide balanced activity against Gram-positive and Gram-negative organisms, with additional coverage of P. aeruginosa. Unlike previous generation cephalosporins, they are bestowed with high-affinity binding to multiple penicillin-binding proteins (PBPs) and relative stability toward several β-lactamases.19,21 For instance, cefepime and cefpirome are relatively stable to AmpC, Klebsiella pneumoniae carbapenemase (KPC), and metallo-β-lactamase (MBL) expressed in Escherichia coli and Klebsiella pneumoniae. 19
Cefepime and cefpirome also bridge the spectrum gaps of third-generation cephalosporins with improved activity against derepressed AmpC in Enterobacter spp., Citrobacter spp., and Proteus spp. In addition to β-lactamases stability, potent antipseudomonal action makes these agents suitable for the treatment of Gram-negative pneumonia. Good coverage of Gram-positive organisms, particularly penicillinase-producing Staphylococcus aureus turned cefepime into a valuable monotherapy option for febrile neutropenia and in immunosuppressed patients (such as cancer patients). Owing to optimal antimicrobial as well as PK profile coupled with low protein binding-driven vascular and lung concentrations, on-therapy development of resistance for cefepime and cefpirome has rarely been reported.19,22,23
Although, in laboratory-based resistance studies, cefepime and cefpirome have been shown to select outer-membrane porins-mediated resistance, it was rarely observed in clinical situation. Rather, eventual emergence of newer/mutated β-lactamases owing to the excessive use of BL-BLI combinations led to some compromise in their efficacy.24,25 Such newer variants of SHV and TEM and more recently CTX-M, oxacillinases, ultimately led to shifting of therapeutic options from stand-alone cephalosporins to combinations based on BL-BLI such as amoxicillin-clavulanic acid, ampicillin-sulbactam, and piperacillin-tazobactam (PIP-TAZ) as shown in Fig. 1. 26
The BL-BLIs Combinations
Another pragmatic approach of tackling pathogens expressing β-lactamases was combining BLs with β-lactamase inhibitors (BLIs). Combining a BLI with clinically established BL is an attractive strategy as it allows restoring the activity of BL for which significant amount of microbiological, PK, safety, and clinical experience is available. 27 Use of BLIs such as clavulanic acid, tazobactam, and sulbactam helped restore the utility of antibiotics such as amoxicillin, ampicillin, ticarcillin, and piperacillin.
Attempts to discover BLI were initiated in early 1940 and 1950, however, without success. Subsequently, a program of screening of organisms naturally synthesizing BLIs was started after 1967. 28 This strategy gave birth to three BLIs, clavulanic acid, sulbactam, and tazobactam. Eventually, amoxicillin-clavulanate, ticarcillin-clavulanate, ampicillin-sulbactam, and PIP-TAZ received approval for clinical use in the United States and Europe. 27
Amoxicillin-clavulanate
Amoxicillin-clavulanate was introduced in the United Kingdom in 1981 and later in the United States in 1984. 27 Clavulanic acid restores the activity of amoxicillin against penicillinase-producing organisms such as S. aureus, Haemophilus influenzae, Moraxella catarrhalis, Neisseria gonorrhoeae, and few Gram-negative organisms that produce narrow-spectrum class A β-lactamases. 27 This is the only BL-BLI combination that is also available for oral use. The combination is most extensively used for the treatment of respiratory infections in which Streptococcus pneumoniae is a dominant pathogen. However, it should be noted that clavulanic acid does not benefit amoxicillin against S. pneumoniae, as this organism is not known to produce β-lactamases. 29 Even for H. influenzae, the impact of clavulanic acid is limited, as significant proportion of H. influenzae clinical isolates are resistant due to PBP3 modification against which clavulanic acid has no role to play. 30 The high vulnerability of amoxicillin for extended-spectrum β-lactamases such as SHV, TEM, CTX-M, and low dose of clavulanic acid used in the combination makes the combination inappropriate for the treatment of contemporary Gram-negative infections. However, owing to scarcity of effective oral therapies for Gram-negative infections, the amoxicillin-clavulanic acid combination is often used as step-down therapy, possibly leading to emergence of inhibitor-resistant TEM, SHV, and Class C enzymes (Fig. 1).
Clavulanic acid is also combined with ticarcillin, which was the first BL-BLI combination receiving marketing approval for intravenous use. Owing to high dose of ticarcillin (3 g administered four to six times/day) and reasonably good activity against then prevailing P. aeruginosa isolates, the combination found utility for the treatment of infections caused by this organism. 27 However, clavulanic acid is unable to inhibit β-lactamases associated with P. aeruginosa rather acts as an inducer of certain pseudomonal enzymes and, therefore, the antipseudomonal activity of the combination is solely driven by the activity of ticarcillin. 31 Even for infections caused by other Gram-negatives, efficacy of the combination is compromised owing to the lower dose of clavulanic acid used.
Retrospectively, it seems that, the coverage profile of amoxicillin-clavulanate and ticarcillin-clavulanate could have been improved by employing higher dose of clavulanic acid enabling them to cover even contemporary pathogens. However, it is plausible that employing higher dose of clavulanic acid was challenging due to the reported adverse effects associated with clavulanic acid, such as diarrhea, abdominal discomfort, and flatulence. 32 Alternatively, rather than β-lactamase labile penicillins, combining clavulanic acid with relatively more stable cephalosporins marketed then, would have offered more effective combinations than amoxicillin-clavulanate and ticarcillin-clavulanate. Cephalothin, cefazolin, cefadroxil, cephalexin, and cephradine could have been potentially worthy candidates for combination with clavulanic acid.
In the hindsight, it seems that since amoxicillin, ticarcillin, and clavulanic acid, all were discovered at Beecham Pharmaceutical, combining them was more opportune as combining with cephalosporins discovered at other companies. However, this would have entailed overcoming complex patent-related ownership challenges. Furthermore, companies tend to choose their off-patent cephalosporin to combine with new inhibitor as it helps leverage the wealth of information available for the cephalosporin, overall making the combination development somewhat less challenging. Moreover, such combination helps extend the patent life of the off-patent cephalosporin—a commercially alluring prospect for a company. In this regard, it is worth mentioning some of the recent efforts of combining optimal older cephalosporins with older BLIs. Such ingenuity was initially demonstrated by certain Indian pharmaceutical companies that introduced oral combination of clavulanic acid with cefixime, cefpodoxime, and cefadroxil.33,34 Against ESBL-producing strains, cefixime-clavulanic acid and cefpodoxime-clavulanic acid shows significantly improved activity compared with amoxicillin-clavulanic acid.35,36 Recently, Achaogen, Inc. reported the development of ceftibuten-clavulanic acid combination, which has also been shown to possess high degree of activity against ESBL-expressing Enterobacterales. 37
Ampicillin-sulbactam
Ampicillin has been combined with a penicillinate sulfone BLI-sulbactam, and similar to amoxicillin-clavulanate, shows improved activity against β-lactamase-producing H. influenzae, as well as certain other Gram-negative organisms such as E. coli and Proteus spp. Ampicillin-sulbactam provides coverage of a wide spectrum of bacterial pathogens, including Gram-positive and Gram-negative bacteria as well as anaerobic organisms. 38 Thus, the combination is used as a monotherapy in clinical situations requiring the use of multiple antibiotics. 27 Unlike ticarcillin-clavulanate, the combination lacks activity against P. aeruginosa. However, owing to intrinsic antiacinetobacter activity of sulbactam (as well as another penicillinate sulfone BLI-tazobactam), the combination offered a valuable option for the treatment of Acinetobacter baumannii infections. Anti-acinetobacter activity of the sulbactam is driven by several interesting attributes not available with clavulanic acid.
In a recent study, it has been demonstrated that sulbactam shows high-affinity binding to PBP3 (IC50: 0.9 μg/mL) and PBP1b (IC50: 0.64 μg/mL) of A. baumannii leading to direct antibacterial activity. 39 Another attractive aspect of sulbactam is its relative stability to several A. baumannii-derived oxacillinases as well as acinetobacter-derived cephalosporinases (ADCs) such as ADC-7.39–41 Owing to high-dose safety, the sulbactam/ampicillin-sulbactam has often been used at high doses for the treatment of A. baumannii infections. 42 However, the activity of sulbactam/ampicillin-sulbactam is significantly compromised against carbapenem-hydrolyzing oxacillinases in A. baumannii, which are now widely prevalent all over the world.
In Enterobacterales, high level of class C β-lactamase expression, and/or low outer membrane permeability for ampicillin may override the capacity of sulbactam in inhibiting β-lactamases. The resistance to this combination has been ascribed to overexpression of TEMs pointing toward poor enzyme inhibition profile of sulbactam and high enzymatic susceptibility of ampicillin.27,43 Even though the dose of sulbactam employed in the combination is quite high, the poor PK features driven by higher protein binding (38%) results in suboptimal coverage of β-lactamase-producing Gram-negative pathogens. 44 Thus, similar to the case of clavulanic acid, a combination of sulbactam with a relatively more β-lactamase stable cephalosporin could have offered more effective combination. However, owing to the ownership issues, the development of ampicillin–sulbactam combination was more opportune as they both belonged to Pfizer.
Piperacillin-tazobactam
PIP-TAZ is characterized by its activity against P. aeruginosa, ESBL-expressing Enterobacterales, and anaerobes.43,45,46 The combination turned out to be very successful as first-line empiric therapy for the treatment of ESBL-producing Enterobacterales and P. aeruginosa.47–49 Piperacillin is bestowed with interesting features of high-affinity Gram-negative PBP binding and better stability to TEMs as compared with other penicillins.50,51 Favorable safety profile facilitated the use of this agent at high doses that helped provide therapeutically relevant exposures against ESBL-producing pathogens. However, piperacillin is highly labile to hydrolysis particularly in the event of concurrent expression of ESBLs and class C β-lactamases.52,53 Since tazobactam is a modest inhibitor of class C enzymes, the doses used in combination with piperacillin are not adequate in tackling class C-expressing pathogens. 54 Thus, with the increase in the numbers of multiple ESBL, and ESBL + AmpC/class C-expressing contemporary pathogens, the PIP-TAZ combination suffered efficacy erosion (Fig. 1).55–57
This was revealed in a recent multicenter “MERINO” clinical trial involving patients with bloodstream infection (BSI), which showed higher mortality in PIP-TAZ arm as compared with meropenem arm even though the infecting pathogens in both the arms were susceptible to respective drug. 58 With respect to P. aeruginosa, similar to clavulanic acid, tazobactam does not improve the activity of partner penicillin against cephalosporinase-producing strains. 59 This could partly be due to suboptimal permeation of tazobactam in this pathogen possibly due to low plasma concentrations attained with 500 mg dose of tazobactam in PIP-TAZ. 60 Thus, selected clinical doses of PIP-TAZ are not able to cope up with contemporary highly resistant pathogens.
In this context, combining tazobactam with relatively more β-lactamase stable cephalosporin would have certainly yielded a superior combination product. However, since both the constituents of the combination PIP-TAZ were owned by Wyeth, combining them offered relative ease of development and extended patent protection to piperacillin. Studies have shown that against multiple ESBL and/or class C-expressing strains, lower MICs are obtained when cefepime or cefpirome is combined with tazobactam as compared with the MICs of PIP-TAZ combination. This is due to higher enzymatic stability of these fourth-generation cephalosporins for contemporary β-lactamases as compared with piperacillin. 61 Mechanism of action studies show that cefepime is a better cephalosporin than cefpirome as the former is uniquely bestowed with high-affinity binding to PBP2 (in addition to PBP3) imparting it a rapid bactericidal action.21,62 Recently reported high-dose cefepime-tazobactam combination (WCK 4282) is, therefore, able to comprehensively overcome PIP-TAZ resistance in MDREnterobacterales. 63
Carbapenems
Carbapenems were true advancements in the management of Gram-negative infections, due to their unique pharmacophore rendering them stability to ESBLs, class C, and AmpC β-lactamases. 64 As a result, carbapenems are considered one of the most reliable drugs for treating serious Gram-negative infections. Mechanistically, they are differentiated from other BLs in terms of possessing high-affinity PBP2 (IC50: 0.008 μg/mL) binding in addition to PBP3 (IC50 range: 0.6–8 μg/mL) or PBP1a/b (IC50 range: 0.4–1.7 μg/mL). Thus, they inhibit majority of essential PBPs with high affinity and, therefore, induce rapid bactericidal action.65–67 Such bactericidal action confers a significant PK/PD gain to carbapenems, which is manifested in terms of lower requirement of free T>MIC as compared with cephalosporins and penicillins.65–67 Carbapenems overall possess potent activity toward all three major Gram-negative pathogen group—Enterobacterales, P. aeruginosa, and A. baumannii. 68 In particular, meropenem is more active against P. aeruginosa as compared with imipenem and, therefore, is a preferred therapy option for P. aeruginosa-dominated indications such as hospital-acquired pneumonia/ventilator-acquired bacterial pneumonia (HABP/VABP).69,70 Even though doripenem shows a slight advantage over meropenem in terms of antipseudomonal potency, its failure in VABP clinical study is a reflection of its relatively suboptimal PK/PD owing to the selection of lower dose (doripenem: 500 mg, q8h; meropenem: 1/2g q8h).71,72
Unlike other carbapenems, ertapenem possesses longer half-life of 4 hours possibly a reflection of high protein binding (∼85%–95%), hence is administered as a once-a-day drug that is suitable in outpatient parenteral antimicrobial therapy settings.73,74 Although ertapenem demonstrates excellent activity against ESBL and Class C/AmpC-expressing Enterobacterales but is impacted by impermeability and also lacks clinically relevant activity against P. aeruginosa. 75 Thus, among carbapenems, meropenem offers a superior therapeutic profile for the treatment of infections caused by P. aeruginosa due to the availability of high doses, flexibility of administering longer duration of infusion, and good track record in the treatment of HABP/VABP. 60
Imipenem, in contrast, demonstrates excellent activity against Enterobacterales, however, is adversely impacted by impermeability-based mechanism of resistance often expressed in P. aeruginosa. 76 However, since imipenem was introduced much earlier in clinical practice, it was extensively used in serious infections caused by Enterobacterales as well as P. aeruginosa leading to incidences of on-therapy resistance development particularly for P. aeruginosa.77,78 For another challenging nonfermenter, A. baumannii, carbapenems and sulbactam-based combinations offered valuable therapeutic options. 79 In particular, for ADCs expressing A. baumannii, carbapenems owing to their rapid cidal action offer a better therapeutic option as compared with sulbactam-based combination.80,81
In addition, carbapenems are characterized by optimal PBP binding profile in A. baumannii and good permeation features in A. baumannii.81,82 Both imipenem and meropenem exhibit similar PBP binding profile in A. baumannii; however, unlike imipenem, meropenem offers advantages of higher doses, potential to extend infusion and relatively ease of dose adjustment in patients with renal impairment. Before the approval of meropenem, imipenem was extensively used to manage A. baumannii infections possibly leading to evolution of carbapenem-hydrolyzing oxacillinases such as OXA-23, OXA-24, OXA-27, and OXA-27. Acquisition of additional resistance mechanisms such as downregulation of porins such as OccAB1-OccAB5, OprD, CarO, and Omp25 and hyperexpression of AdeABC efflux pumps transformed this pathogen as one of the most challenging organism ultimately leading Centers for Disease Control and Prevention and World Health Organization listing this pathogen under “serious threats” and “critical priority” categories, respectively.83–87
In Enterobacterales, initial carbapenem resistance was mainly associated with strains expressing AmpC in conjunction with impermeability. Furthermore, investigations revealed that in relative terms, imipenem is more vulnerable to hydrolysis by AmpC, particularly in strains co-expressing impermeability.88,89 Meropenem, in contrast, is quite stable to AmpC or class C β-lactamases but is vulnerable to class A ESBLs when expressed in Enterobacterales with porin loss.90,91 However, unlike imipenem, on-therapy resistance development has not been frequently reported for meropenem, which could be ascribed to the availability of high-dose/extended infusion for meropenem. 92 This point toward the clinical advantages associated with high-dose safety of a given drug. As carbapenems emerged as reliable therapy options for hospital infections, their extensive use led to emergence and spread of carbapenem-resistant Enterobacterales (CREs) such as KPC, MBL, and OXA-48/181 like as well as OXA-232 (Fig. 1).
Earliest carbapenem resistance was found in Bacillus cereus, which was due to chromosomally encoded carbapenemases that had low transferability. 93 However, in Gram-negatives, resistance mechanisms such as NDM, KPC, and OXA-48/181 are plasmid borne leading to their extensive dissemination in Enterobacterales. 94 Therefore, limiting the use of imipenem for the treatment of infections caused by Enterobacterales and meropenem for infections caused by P. aeruginosa could have possibly reduced selection pressure leading to CREs. Such a scenario points toward the need of availability of multiple therapeutic options to tackle each pathogen/resistance mechanisms so that clinicians are at a liberty to select the most appropriate one with the objective of maximizing therapeutic success and curtailing resistance development.
From the previous commentary, it is clear that all the older BL-BLI combination have lost significant degree of efficacy against contemporary pathogens and increased use of carbapenems have exerted significant selection pressure leading to emergence and spread of CREs. As a result, the clinical community is facing a severe dearth of reliable empiric therapy options that would minimize CRE selection pressure thereby aiding antibiotic stewardship programs. International discovery teams responded to this grave situation and directed their efforts toward discovering novel BL-BLI combinations. These efforts led to the successful introduction of ceftazidime-avibactam, ceftolozane-tazobactam, meropenem-vaborbactam (MEM-VAB), and recently, imipenem-relebactam.
Among late-stage BL-based products, we focus on two cefepime-based therapeutically well-demarcated products, one with potential to serve as first-line carbapenem-sparing and PIP-TAZ replacing therapy for the treatment of infections caused by contemporary Gram-negative pathogens (WCK 4282, high-dose extended infusion cefepime-tazobactam) and the other potentially addressing the need for the ultimate second- or third-line “destination therapy” for MDR/XDR pathogens and thus supplanting colistin (WCK 5222, cefepime-zidebactam). Extensive microbiological and PK/PD-based information has been published for both the combinations that facilitate drawing inference on their potential clinical role upon successful development. We also share an overview of monocyclic BL-based combination—aztreonam + avibactam under late-stage development targeted at MBL-expressing pathogens.
Newer BL-BLI Combinations
Ceftazidime-avibactam
Ceftazidime, an established third-generation cephalosporin, has been combined with avibactam a novel diazabicyclooctane inhibitor originally discovered at Aventis (also known as AVE 1330a) with expanded BLI spectrum. Unlike older BLIs with BLI spectrum restricted to class A enzymes, avibactam is able to inhibit class C β-lactamases as well as KPCs that are widely reported from the United States, Europe, and China with the exception of some countries such as India.95–99 Avibactam shows a well-differentiated mechanism of BLI as unlike older inhibitors avibactam is deacylated and released from the enzyme, thus regenerating the intact avibactam that can now again undergoes acylation by other, or the same, β-lactamases. 97 This is unlike previous BLIs that follow hydrolytic path yielding clipped molecules without inhibitory activity. Ceftazidime-avibactam demonstrates potent activity against strains expressing ESBL, class C, mixed enzyme, KPC, and OXA-48/181 (including strains with impermeability).97,100,101
For KPC-expressing pathogens, ceftazidime-avibactam offers a unique therapeutic option as none of the previous BL-BLI combinations possessed meaningful activity against KPC-producing organisms. The combination also has a clinically valuable activity against ceftazidime-nonsusceptible P. aeruginosa, as avibactam is a good inhibitor of pseudomonas-derived cephalosporinases, however, shows limited activity against VEB and OXA-expressing as well as carbapenem-resistant P. aeruginosa.102–104 Ceftazidime-avibactam combination was initially approved for the treatment of complicated UTI (cUTI) and complicated IAI (cIAI), and recently gained approval for HABP/VABP (U.S. Food and Drug Administration label).
Disturbingly, early reports of on-therapy resistance development in pneumonia and BSI patients re-emphasize the fact that Gram-negative pathogens are extremely versatile with an enormous potential to escape the effects of newer therapies.105–107 It has been reported that mutations in Ω-loop of KPC results in enhanced affinity toward ceftazidime resulting into impaired inhibition of KPC by avibactam. Certain studies also postulate that single amino substitution in KPC-2 (D179Y) is sufficient to prevent its inhibition by avibactam leading to resistance to the ceftazidime-avibactam combination. 108 To complicate the matter even more, recent studies have also shown mutations in AmpC, ESBL, and OXA-48 as a result of ceftazidime-avibactam exposures.109–111 Furthermore, the resistant mutants have also been demonstrated to acquire nonenzymatic resistance mechanism such as impermeability. 107
Unleashing of multiple mechanisms of resistance by Enterobacterales against ceftazidime-avibactam within 5 years of its approval re-emphasizes the concurrent availability of multiple therapeutic options effective against older as well as newer resistance mechanisms. From the PK/PD perspective, possibly, the resistance development against ceftazidime-avibactam could have been diminished by employing higher doses of avibactam and/or selecting cefepime as the partner drug that has obvious mechanistic advantages over ceftazidime as discussed in the previous sections. 112 However, a slight potency advantage against P. aeruginosa, acceptability in larger number of countries and early out-of-patent availability might have favored the selection of ceftazidime. If continued unabated, resistance development against ceftazidime-avibactam could lead to loss of this valuable sole cephalosporin-based therapy for infections caused by KPC organisms. Therefore, use of ceftazidime-avibactam in cUTI with suspected or documented involvement of class C or KPC-expressing pathogen would not only ensure good clinical outcome but would also minimize the propensity of resistance development. Assurance of clinical efficacy in cUTI emanates from availability of high concentrations of both ceftazidime and avibactam in renal tissues, as both undergo urinary elimination. Moreover, use of ceftazidime-avibactam should be discouraged in regions reporting high level of carbapenem resistance in P. aeruginosa and Enterobacterales.
Ceftolozane-tazobactam
Ceftolozane was originally discovered by a Japanese company Astellas, which was later on licensed to Calixa, which in turn was taken over by Cubist, and finally landed with Merck as a result of Merck acquisition of Cubist. Ceftolozane has a special feature of potent activity against MDR P. aeruginosa owing to its stability toward pseudomonal AmpC and minimal impact of pseudomonal efflux.113,114 However, ceftolozane-tazobactam shows limited activity against VEB and OXA-expressing P. aeruginosa.15,115 It has been reported that tazobactam does not help further improve the activity of ceftolozane against P. aeruginosa. 116 From the angle of empiric use potential, the combination has poor coverage of ESBL-expressing Enterobacterales owing to extremely labile nature of ceftolozane for ESBL and class C enzymes and lower doses of tazobactam used in the combination. 116 Ceftolozane-tazobactam is also inactive against KPC as well as OXA-181-expressing organisms that invariably co-express ESBL/class C enzymes. 116 Therefore, to ensure favorable efficacy use of ceftolozane-tazobactam needs to be restricted for the treatment of documented P. aeruginosa infections and should not be employed as an empiric therapy for the treatment of infections caused by ESBL-expressing Enterobacterales.
Meropenem-vaborbactam
MEM-VAB is a recently approved BL-BLI combination. This combination is based on vaborbactam, which is a boronate-based new chemical entity. 117 From the microbiology point of view its activity profile is just similar to ceftazidime-avibactam with a disadvantage of lack of coverage of OXA-48/181 expressing Enterobacterales.117,118 However, MEM-VAB has been shown to be active against ceftazidime-avibactam-resistant KPC-producing K. pneumoniae, which could be clinically valuable feature in view of several incidences of resistance development for ceftazidime-avibactam resistance in KPC-expressing pathogens. 119 Use of high dose of meropenem brings in comprehensive coverage of ESBL and class C-expressing pathogens; however, use of this agent in empiric setting is likely to augment the dissemination of CREs for which therapeutic options are extremely limited. Moreover, this combination would be challenged by meropenem-impacting nonenzymatic resistance mechanisms such as efflux and impermeability as vaborbactam does not help tackle such resistance mechanisms. 120 This is a significant limitation as prevalence of efflux-based mechanism of resistance in P. aeruginosa is fairly high.121,122 Furthermore, particularly against P. aeruginosa, vaborbactam does not help improve the activity of meropenem.120,123 From clinical use point of view, the use of MEM-VAB should be restricted in regions reporting high prevalence of class D carbapenemase-expressing Enterobacterales.
Imipenem-relebactam
Imipenem-relebactam is the most recently approved combination and demonstrates a coverage profile similar to MEM-VAB with OXA-48/181 expressing pathogens being beyond the therapeutic scope of this combination as well.124,125 Nevertheless, the combination demonstrates good activity against P. aeruginosa expressing derepressed AmpC with or without OprD downregulation and efflux as well as OXA-expressing phenotypes. 126 Therefore, the combination may find useful clinical utility for the treatment of pseudomonal infections, however, should not be used in infections suspected to involve Enterobacterales expressing OXA-48/181-like pathogens or P. aeruginosa expressing carbapenem resistance.
Although aforementioned carbapenem-based therapies indeed offer additional options for the treatment of KPC infections, past clinical experience pointing toward a definitive link between carbapenem use and emergence of resistance indicates the potential risk of CRE dissemination due to the selection pressure exerted by such agents.
Aztreonam-avibactam
As discussed, increased usage of carbapenems has led to emergence of diverse carbapenem-impacting resistance mechanisms with MBL being in the forefront of therapeutic challenge. To address this menace a combination of aztreonam and avibactam is under development. The combination is based on the principle of using an MBL stable monocyclic BL—aztreonam combined with broad-spectrum inhibitor of class A and C enzymes, avibactam. Successful development of the combination would potentially provide an effective therapy against Enterobacterales expressing MBLs in conjunction with other β-lactamases. 127 However, aztreonam-avibactam is marred by a clinically important spectrum gap as it is unable to cover a recently reported resistance mechanism involving PBP3 modifications in E. coli. Multiple reports have identified a four amino acid insertions (YRIK or YRIN) in PBP3 in clinical isolates of E. coli that hampers the binding of aztreonam to its prime target PBP3. 128 Against such strains, the MICs of aztreonam-avibactam combination typically rise beyond 8–16 mg/L, possibly beyond the therapeutic scope of this combination.128–130 The challenge posed by this resistance mechanism is aggravated with the reports of PBP3 inserts in MBL as well as ESBL expressing E. coli. In addition, the major pathogen spectrum gaps in this combination are P. aeruginosa and A. baumannii that would preclude the use of this agent for difficult-to-treat indications such as HABP/VABP and BSI.127,131
WCK 4282 (high dose, extended infusion, cefepime-tazobactam)
WCK 4282 is a combination of high-dose cefepime and tazobactam (2 + 2 g) to be administered three times a day with extended infusion (1.5 hours). This combination is at phase 3 stage of clinical development and the published literature suggests its potential therapeutic role as a first-line empiric use therapy. The antimicrobial activity of WCK 4282 has been demonstrated to be comparable with carbapenems thereby presenting a carbapenem sparing therapeutic option—a much needed feature supporting antibiotic stewardship and minimizing the emergence of CRE. 132 WCK 4282 shows potent activity against Enterobacterales that co-express multiple ESBLs and class C enzymes that are invariably nonsusceptible to PIP-TAZ, cefoperazone-sulbactam and even ceftolozane-tazobactam.132–134 Owing to compromise in efficacy of PIP-TAZ for the treatment of infections caused by multiple ESBL/class C-expressing pathogens, upon successful development, WCK 4282 would represent a superior therapeutic option. The combination involves the use of highest ever dose of tazobactam that facilitates effective sparing of relatively β-lactamase stable cefepime, from the hydrolytic attack by multiple ESBLs, ESBL + class C, and KPCs expressing pathogens. Furthermore, the selected dose of WCK 4282 provides potential coverage of OXA-48/181 expressing isolates as well.133,135
Owing to such features, WCK 4282 has a distinct potential to emerge as a superior alternative to PIP-TAZ, ceftolozane-tazobactam and cefoperazone-sulbactam thus providing a “true” first-line empiric therapy for contemporary Gram-negative infections. This would minimize the dependence on carbapenems and reinstate their erstwhile role as last resort effective therapies for ESBL infections. However, upon successful approval, the use of this combination needs to be avoided in high carbapenem-resistance settings.
WCK 5222 (cefepime-zidebactam)
The approach of BLI-based therapies did serve well in the past; however, in the present circumstances of multiplicity of ever-evolving resistance mechanisms, it is extremely challenging to discover a BLI with ability to inhibit all the four classes of β-lactamases as well as tackle nonenzymatic resistance mechanisms. Moreover, ongoing emergence of newer β-lactamases and inhibitor-resistant mutant β-lactamases would continue to challenge the BLI-based approach. To overcome all these resistance mechanisms, a novel approach based on β-lactam enhancer has been recently proposed.
WCK 5222 is a novel mechanism of action-based combination of cefepime and a high-affinity PBP2 binding β-lactam enhancer antibiotic, zidebactam. Cefepime-zidebactam is at phase 3 stage of clinical development. The β-lactam enhancer-based mechanism action of this combination exerts activity against all the known resistance mechanisms in Gram-negatives, including CREs, P. aeruginosa, including MBL and class D expressing A. baumannii.125,136,137 The zidebactam's β-lactam enhancer action obviates the need of BLI and, therefore, overcomes the resistance mediated through all the four classes of β-lactamases. 138
Published reports indicate that unlike other BL-BLI therapies wherein a BLI merely restores the activity of BL, in WCK 5222, zidebactam augments the bactericidal activity of cefepime. 138 Enhancement of cefepime activity by zidebactam leads to lowering of cefepime's %T>MIC requirement, a PD gain facilitating coverage of high MIC MDR/XDR pathogens. Such enhanced bactericidal action of WCK 5222 is expected to provide reassuring efficacy in most difficult-to-treat patient population inflicted with HABP/VABP and BSI. Thus, published in vitro, in vivo, and clinical studies support WCK 5222's potential colistin-supplanting agent for the treatment of serious infections with limited or no treatment options.
Based on the previous commentary it seems that it is important to identify appropriate clinical situations for which a specific Gram-negative therapy needs to be deployed based on their PK/PD and various microbiological aspects. Figure 2 depicts the desirable features in empiric and definitive therapies based on the pathogen/resistance coverage spectrum. For example, an empiric therapy is expected to provide a broad-spectrum coverage against commonly encountered Gram-negative pathogens such as E. coli, K. pneumoniae, Enterobacter spp., Serratia spp., and P. aeruginosa. Considering widespread prevalence of ESBLs, AmpC + ESBLs, ESBLs + OXA, and E. coli with PBP3 inserts as well as ESBLs + OprD + AmpC in P. aeruginosa, an empiric therapy needs to cover these resistance mechanisms. However, as shown in Fig. 2, therapeutic gaps of these formerly listed BL-BLI combinations such as poor activity of PIP-TAZ against Enterobacterales isolates expressing multiple ESBLs + OXA or ESBLs + class C/AmpC enzymes should also be taken in to account before deploying these empiric therapies. This would likely help preserve the therapeutic utility of current and newer BL-based Gram-negative therapies.

Expected antimicrobial profile for empiric and definitive β-lactam-based therapy based on pathogens/resistance mechanisms coverage. MDR, multidrug resistance.
Lessons Learned and Way Forward
During the past 50 years, the pattern of use of antibiotics in empiric settings does not seem to be attuned with their microbiological and PK/PD properties, which led to widespread resistance development resulting into rapid loss of valuable BL-based therapeutic options. Lack of clarity on coverage spectrum, differences in mechanism of action, and PKs often led to the interchangeable use of BL agents. Importantly, concurrent nonavailability of multiple therapeutic options targeted toward a pathogen/resistance mechanism led to compulsion of using the sole drug option in all the clinical settings, including efficacy-compromising and/or resistance-promoting situations. Therefore, it needs to be re-emphasized that discovery and development of novel antibacterial agents must be an ongoing endeavor and should not rest on past laurels, undermining the versatility of pathogens in deploying smart evasive novel mechanisms of resistance. On a brighter note, several novel BL-BLI combinations have been recently approved, whereas some are at an advanced stage of clinical development. To be able to retain their effectiveness for a longer duration, it is critical to comprehend their clinically relevant microbiological, mechanistic, and PK/PD features for deploying them in appropriate clinical situations.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received.