
Abstract
Acinetobacter baumannii is known as a Gram-negative bacterium that has become one of the most important health problems due to antibiotic resistance. Today, numerous efforts are being made to find new antibiotics against this nosocomial pathogen. As an alternative solution, finding bacterial target(s), necessary for survival and spread of most resistant strains, can be a benefit exploited in drug and vaccine design. In this study, a list of extensive drug-resistant and carbapenem-resistant (multidrug resistant) A. bumannii strains with complete sequencing of genome were prepared and common hypothetical proteins (HPs) composed of more than 200 amino acids were selected. Then, a number of bioinformatics tools were combined for functional assignments of HPs using their sequence. Overall, among 18 in silico investigated proteins, the results showed that 7 proteins implicated in transcriptional regulation, pilus assembly, protein catabolism, fatty acid biosynthesis, adhesion, urea catalysis, and hydrolysis of phosphate monoesters have theoretical potential of involvement in successful survival and pathogenesis of A. baumannii. In addition, immunological analyses with prediction softwares indicated 4 HPs to be probable vaccine candidates. The outcome of this work will be helpful to find novel vaccine design candidates and therapeutic targets for A. baumannii through experimental investigations.
Introduction
A
Over the last two decades, dissemination of multidrug-resistant (MDR), extensive drug-resistant (XDR), and pan-drug-resistant A. baumannii strains has produced therapeutic challenges and consequently increased mortality of patients. 8 A. baumannii strains resistant to all clinically used antibiotics against the Gram-negative bacilli have been already reported. 8 So, existing ineffective treatments should be replaced with new therapeutic and preventative strategies. Vaccination is also a therapeutic approach, decreasing morbidity and mortality of patients as well as necessity of their antibiotic use. 9 Therefore, main virulence factors (VFs) of A. baumannii should be identified for successful construction of vaccines and drugs. To date, a few VFs such as lipopolysaccharide (LPS), 10 capsular polysaccharides, 11 biofilm-associated protein (BAP), 12 outer membrane protein A (OmpA), 13 penicillin-binding protein 7/8, 14 surface autotransporter (Ata), 15 surface polysaccharide poly-N-acetyl-β-(1–6)-glucosamine (PNAG), 15 and AbaI autoinducer synthase 16 have been attributed to A. baumannii.
Efficiency of some of these antigens, as vaccine candidates and drug targets, has been also investigated in preclinical studies in animal models. Some of these candidates possess high vaccination potential. However, some drawbacks are obvious in these studies. Some of these problems are related to antigen characteristics such as its variability and solubility 17 and others are about preparation and administration methods like high costs of molecules purification, low stability of purified antigens, and limited protection results. 18
A “hypothetical protein” (HP) is one that is estimated to be encoded by an open reading frame, but no protein product has been characterized by experimental methods. Since these proteins are abundant and they may play important roles in survival and virulence of a pathogen, there is a clear need for functional annotation of this kind of protein.19,20 A precise identification and characterization of HPs from several important human pathogens using in silico methods have been also reported.21–25 The already-sequenced genomes of antibiotic-resistant and hypervirulent A. baumannii strains were taken in this study to explore the precise function of their common HPs using bioinformatic tools. Afterward, probable potent targets for vaccine and drug design studies were introduced.
Methods
Sequence retrieval
The list of A. baumannii strains with finished genome sequencing was extracted using the “Phylogenetic Profiler” option under the “Find Genes” section of Integrated Microbial Genome site. 26 Then the strains introduced as hypervirulent, carbapenem resistant (MDR), and XDR in the related literature were determined (Table 1). In the next step, A. baumannii strain LAC-4, as an important hypervirulent and outbreak-associated strain, 27 was selected as a query and its common homologous protein-encoding sequences in other strains listed in Table 2, absent in nonpathogenic Acinetobacter baylyi and human, were retrieved. After exclusion of pseudogenes, HP sequences with a length ≥200 amino acids (aa) were selected as the final dataset.
Selected Hypervirulent, Carbapenem-Resistant (Multidrug Resistant), and Extensive Drug-Resistant Strains of Acinetobacter baumannii
MDR, multidrug resistant; XDR, extensive drug resistant.
The gene Identifiers of 18 Common Proteins Among Hypervirulent and Antibiotic-Resistant Strains of Acinetobacter baumannii with a Length of More Than 200 Amino Acids
HP, hypothetical protein.
Determination of codon adaptation index
The expression probability of HP sequences was revealed by calculation of codon adaptation index (CAI) using CAIcal tool. 28 The predicted value was then compared with the CAI of six housekeeping genes of A. bumannii (e.g., recA, cpn6, gltA, gyrB, gdhb, and gpi).
Primary structure analyses
Theoretical measurements of physicochemical parameters of selected HPs were predicted using Expasy's ProtParam server. 29 The parameters computed by this online server include molecular mass, theoretical isoelectric point (pI), extinction coefficient, 30 instability index, 31 aliphatic index, 32 and grand average of hydropathicity (GRAVY). 33 Estimation of subcellular localization also has great importance in predicting its function. 34 Online tools like CELLO (v2.5), 35 PSLpred, 36 and Gneg-mPLoc 37 have been similarly used for prediction of subcellular localization of proteins. CELLO using multilayered support vector machines (SVMs) is employed to generate prediction results with overall prediction accuracy of 89%. 38 PSLpred is also an SVM-based predictor with an overall high accuracy of 91.2%, which is only utilized to localize proteins in Gram-negative bacteria. 36 Moreover, Gneg-mPLoc is a flexible and powerful predictor that identifies Gram-negative bacterial proteins even if query protein simultaneously exists in more than one subcellular location. 37 Presence of signal peptide cleavage sites was checked by SignalP 4.1 server. 39 This server uses Neural Networks (NN) and Hidden Markov Models (HMM) and is regarded as the most accurate program that analyses N terminal of protein sequences. 40 Involvement of HPs in signal peptide independent secretion, that is, nonclassical protein secretion, was also predicted using SecretomeP 2.0. It is the only prediction tool for nonclassical secretion pathway in bacteria and mammals. 41 To identify the membrane proteins and to predict probable transmembrane helices in HPs, TMHMM242 and HMMTOP 43 servers were employed. Both of these servers are based on an HMM. TMHMM2 demonstrates 97–98% accuracy for transmembrane predictions. 44
Functional analyses
Sequence and structure comparisons
The primary step toward functional annotations of proteins is sequence similarity search in various protein databases. Popular BLAST-P tool 45 was thus used to identify functional homologues in nonredundant (nr) database of proteins. Results with low sequence identities (<25%), low query coverage (<50%), and big E-values (>0.0001) were referred to as remote homologs and excluded. Hits with the highest value of these parameters were also considered probable functional homologs of HPs. The three-dimensional (3D) structure of a protein not only affects its shape and size but also its function. 46 Also, phyre247 was used to find structural homologs of HPs. The results with a confidence level (>50%) and coverage rate (>30%) were considered close homologs.
Domain assignment
Classification of protein on the basis of domain architecture has become the basis of developing various databases of protein families. Thus, simple modular architecture research tool (SMART) was used to identify and analyze protein domains. Synchronization of this online tool with Ensembl, UniProt, and search tool for retrieval of interacting genes (STRING) has increased accuracy of protein domain annotation and architecture analysis. 48
VF analysis
Severity of bacterial infections is due to various VFs produced by pathogens. Therefore, their identification can serve as suitable therapeutic targets. 49 For this purpose, SVM-based VICMpred was used to sort proteins in one of four functional groups, including VF, metabolism, as well as cellular and storage processes. The overall accuracy of this server is 70.75%. 50 DBETH server was also used to predict bacterial exotoxins for humans. The overall accuracy of this server is more than 90%. 51 Furthermore, comprehensive antibiotic resistance database (CARD) was searched for the identification of HPs involved in antibiotic resistance. A highly developed Antibiotic Resistance Ontology (ARO) at the core of the CARD has made it possible to classify the antibiotic resistance gene data. 52 The search results with E-value (>0.0001) was considered a significant match for the given HP.
Functional protein association networks
Protein-protein interactions (PPI) are essential for proper completion of any cellular process. Therefore, their revelation can be beneficial in predicting the function of HPs. 53 Integrative interaction database of STRING (v. 10.5) 54 was thus used to predict possible presence of HPs in interaction network with high confidence. The validation of PPI networks was also performed in the Cytoscape 3.6.1 program. 55
Immunological analyses
Similarity of vaccine candidate HPs to proteins of healthy human gut flora was searched through BLASTp against the proteome of 95 main gut microbiota species. 56 Results with sequence identities (>25%), query coverage (>50%), and E-values (<0.0001) were considered homologs. Allergenic potential of HPs identified as vaccine candidates was predicted by Algpred server prediction. 57 Sensitivity of this prediction server is 95%. 58 Antigenic determinants of these sequences were analyzed using Bioinformatic Antigenic Peptide Prediction (BPAP) tool. The reported accuracy of this method is about 75%. 59 The VaxiJen v2.0 server was used to predict that selected HPs were protective antigen or not. 60 Prediction accuracy of this server is about 82% for bacterial species. 61 Sequences were subjected to B cell epitope prediction using ABCpred. The accuracy of ABCpred in predicting epitopes is 65.93%. 62
MHC class I and class II binding peptides were detected by ProPredI 63 and ProPred 64 servers, respectively. The half maximal (50%) inhibitory concentration (IC50) of common epitopes of both servers was calculated using quantitative prediction of binding affinity of peptide-MHC binding site, MHCPred. 65 Epitopes with IC50 value <100 nM for allele DRB1*0101 were selected as a strong binder epitope.
Results and Discussion
Sequence retrieval
Comparison of genomic sequences of hypervirulent and antibiotic-resistant strains of A. baumannii revealed 118 common HPs, which did not present either in nonpathogenic strains or humans. Among these common proteins, 18 HPs with length more than 200 aa were screened out and selected for further analyses. The identifier of the genes encoding these proteins on the Integrated Microbial Genome Site is presented in Table 2. In the following sections, HP1 to HP18 are used to introduce proteins instead of these identifiers.
Considering that amino acid sequence and total length of a protein influence 3D structure of protein, which in turn impresses protein function, 66 and given that short-length proteins fail to fold properly because of inadequate number of contacts between residues, 67 longer proteins have more chance of accommodating multiple secondary and tertiary structures. Therefore, HP sequences with a length ≥200 amino acids were selected as final dataset in this study.
Codon adaptation index
Since a large number of DNA sequences are registered in the databases, there are numerous statistical procedures to analyze the sequences, including CAI as an interesting one. It soon becomes apparent that there is considerable heterogeneity in codon adaptation among genes of various species. The rate of this index is directly related to the amount of gene expression and also varies from 0 to 1, with a numeric index of >0.5 indicating a high probability of the gene expression. 68 The CAIcal values of the desired HPs in this study were all found to be >0.66. Once this value was compared with the CAI of housekeeping genes of A. baumannii (>0.73) (Table 3), they all had a high probability of expression in A. baumannii strains; so it is worthy to analyze these proteins further.
CAIcal Index Values for Hypothetical Protein Encoding and Housekeeping Genes
CAI, codon adaptation index.
Primary structure analysis
Proteins are continuously synthesized and degraded in cells. The half-life of proteins in bacteria also varies from 20 seconds to 100 days. 69 It should be noted that several factors are involved in the half-life duration of proteins, mainly based on their amino acid composition. One of the destructive pathways of protein is the N-terminal pathway, by which the protein destroyer identifies certain amino acids like leucine (Leu) and phenylalanine (Phe) more than others. 70 According to ProtParam prediction results (Supplementary Table S1), there is only one protein (HP3) with a very short half-life (2 minutes), which contains the leucine amino acid in its N-terminal.
The extinction coefficient also determines how much light is absorbed by a protein at a particular wavelength (280 nm). The presence of the aromatic amino acids of tyrosine and tryptophan in the amount of this absorption is more effective than the formation of sulfide bridges. 71 Presence of aromatic amino acids in the protein structure usually affect folding stability of the protein, association of the protein with other proteins, the protein binding to the ligand, and the protein protection from oxidative damage. 72 All studied HPs have been predicted to have high extinction coefficient (Supplementary Table S1).
The aliphatic index of a protein also shows the amount of occupancy of side chain of proteins with aliphatic amino acids such as alanine, valine, isoleucine, and leucine. Higher numerical value of aliphatic index is thus considered a positive factor in protein temperature resistance. 32 Among the studied HPs, the values of this index are high and vary from 65.25 to 102.09 (Supplementary Table S1).
The pI is the pH at which pure charge of a protein is zero. For a protein with high levels of basic amino acids, pI is high, while an acidic protein possesses lower pI. 73 As shown in Supplementary Table S1, due to the high number of positive-subunit (Arg+Lys) subunits compared with negative-charge subunits (Asp+Glu) in HP2, HP5, HP6, HP16, and HP17, these proteins had higher pI. It was shown that the charge of protein was related to its the location in the cell. It should be noted that cytoplasmic proteins are usually more acidic than those located in the membrane, and DNA-related proteins can be thus acidic or basic. The relatively high pI values of membrane-related proteins are that the positive charges of these proteins can better interact with the negative charges of the polar groups of the phospholipid heads. 74 The low pI values of some proteins (HP4, HP12, and HP14) with predicted membrane-bound localization (Table 4) could be because of their possible post-translational modifications and interaction with the membrane through Ca2+. 75
Results of Used Software to Predict Subcellular Localization, Nonclassical Protein Secretion, Signal Peptide, and Membrane Helixes
Considering the fact that proteins with a score above 0.5 in SecretomeP 2.0 server results are able to participate in secretive pathways as well as matching the results of identifying the signal peptide, the nonclassical secretion pathway, predicting the location of the protein, and calculating the membrane helix (Table 4), it was revealed that proteins HP6, HP13, HP14, and HP17 had predicted signal peptide and participation in nonclassical secretion pathways. However, based on output of SecretomeP acquisition of SecP score exceeding the threshold about 0.5 for bacterial protein sequences, it indicates a nonclassically secreted protein when the signal peptide is not predicted at the same time for that protein. In the case of HP2, there was a contradiction in the result of localization obtained from used predictors, wherein the presence of the peptide signal in that case increased the chance of verifying the extracellular outcome.
According to the SMART software results (Supplementary Table S2), HP16 was likely to have a peptide signal with unmentioned E-value, which had not been predicted by SignalP 4.1 software because of its near-but-below-threshold score. This protein showed the ability to enter the nonclassical secretion pathway and localization in the membrane. It should be noted that both signal peptide and membrane helix contain hydrophobic amino acids, and this sometimes causes controversies in the prediction results by different softwares. 76 The presence of helix between membranes confirms the prediction of the location of this protein in the membrane. This was also true for HP14 for which the prediction result showed the presence of signal peptide (Table 4 and Supplementary Table S2) and membrane helix (Table 4). Accordingly, localization results (Table 4) are variable between outer membrane and extracellular space. HP4, HP8, HP12, HP15, and HP18 showed the ability of entrance to nonclassical secretion pathways among which HP4 and HP12 possessed membranes helixes. Therefore, the probability of their placement in the outer membrane after crossing the inner membrane was high. Despite predicting two membrane helixes at N-terminal of HP5, this protein was predicted to be a cytoplasmic one by software. It is noteworthy that membrane proteins are usually evaluated as possible vaccine targets, while cytoplasmic ones can be assumed as drug targets. 49
Functional analyses
Regarding the prediction of the function of proteins and referring to Supplementary Tables S3–S5 and Tables 5 and 6, the following were revealed about the studied HPs. Proteins that are not mentioned in the following discussion appeared to have no functional significance in the cell or importance in the drug or vaccine design according to the results.
Annotated Functions of Hypothetical Proteins Using BLASTp
SDR, short-chain dehydrogenases/reductase.
Function Prediction of Hypothetical Proteins Using VICMpred
HP1 is predicted to be effective in cellular processes and it can also be a transcription regulating protein from Arabinose operon regulatory protein subfamily (AraC). Many transcriptional regulatory proteins in the bacteria are linked to the DNA through helix-turn-helix motifs (HTH). One of the main subfamilies of these proteins is AraC regulating proteins. These subfamilies are mostly positive transcriptional regulators. The length of sequences associated with these proteins is different, but the HTH motif is usually located at the carboxylic end, 77 which is also found in amino acids 211–250 of HP1. ToxT is one of the members of the AraC-XylS family, which activates the transcription of cholera toxin (CTX) and toxin-coregulated pilus (TCP) genes in Vibio cholerae.78,79 This transcription factor is activated with ToxR and TcpP transmembrane proteins and their internal membrane associates known as ToxS and TcpH. Environmental factors such as temperature, bile salts, pH, and osmolarity are involved in activating these proteins. In addition, ToxT activates RNAs, called TarA and TarB, which are involved in bacterial pathogenesis. 80 Recently, efforts have been made to make new compounds inhibiting the activity of this protein as a therapeutic drug. 81 MarA, SoxS, RamA, and Rob proteins in the Enterobacteriaceae and MtrA family of Neisseria gonorrhoeae are also members of this family, which increase the expression of drug effluxing pumps, reduce the expression of purines, and induce multiple drug resistance. 82 This prediction was in parallel with the prediction of a protein cytoplasmic position illustrated in Table 4.
According to the results, HP4 was similar to that of the pilus assembling protein, which was compatible with its localization prediction in the outer membrane and contained membrane helixes (Table 4). The interaction analysis (Supplementary Table S6) also confirmed this role. A. baumannii expresses different types of pathogenic factors. Type IV pili is an extracellular bacterial appendix, and it is often necessary to connect bacterium to the host cells, bacterial movement, horizontal gene transfer, and biofilm production. The system also consists of a cytoplasmic protein (PilB) and an integral membrane protein (PilC), main units called pilin (PilA), and one or more accessory subunits (PilV). Pilins are usually secreted in prepilin format and after losing their N-terminals and methylation by peptidase enzymes called TFPP, they can be placed in the structure of pili.83,84 Some proteins also contribute to this assemblage.
Findings given in Supplementary Table S6 outline HP4 interactions with peptidase enzyme and PilV. Figure 1 also shows high degree (6) to this protein, indicating that the give protein was very likely to be involved in one of the important pathogenic factors, type IV pilus assembly. Considering the prediction of this protein as a pathogenic agent by VICMpred software (Table 6), it is possible to study the available regions of this protein and the extent of the conservation of these areas among several A. baumannii strains to check whether such areas have potential to stimulate the immune system and to design a vaccine or not.
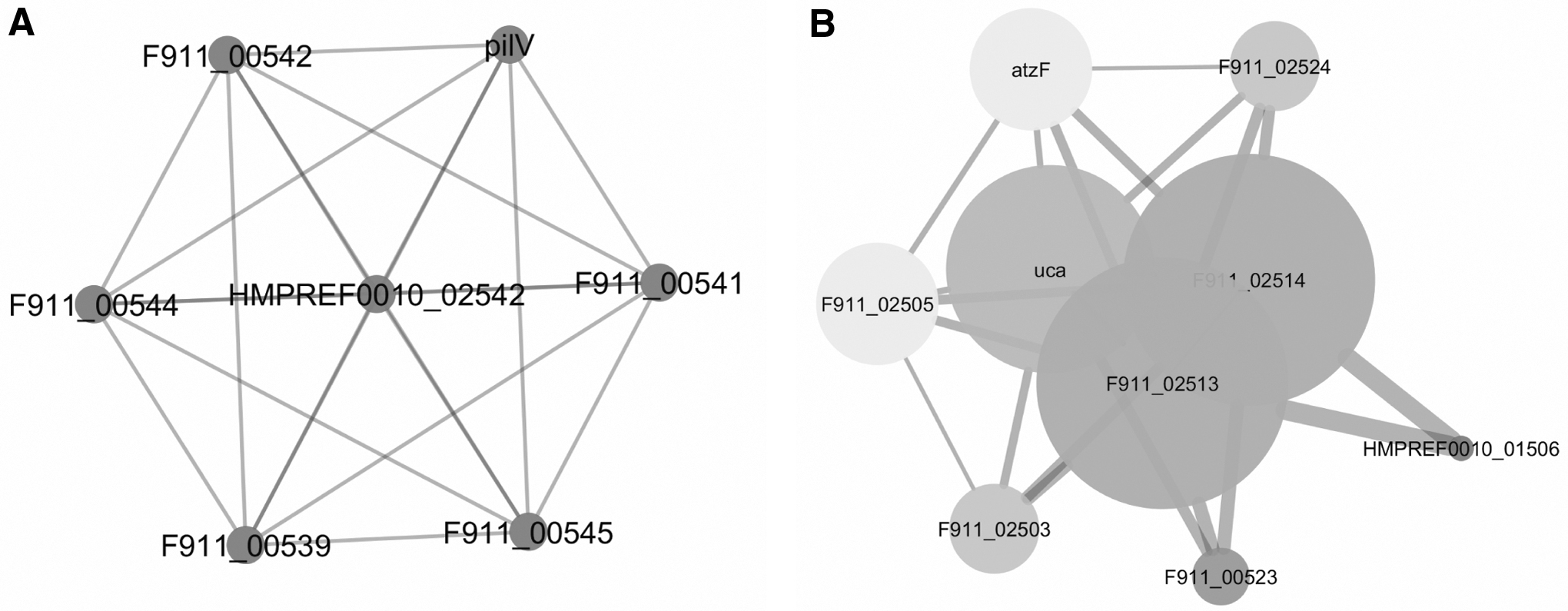
Validation of PPI networks of HP4
Based on the prediction results, HP6 was similar to an ulilysin metalloproteinase. This prediction supported previous discussion of the extracellularity of this protein. Metaloproteases are produced by many pathogenic bacteria and, at the site of the infection, cause cartilage and bleeding tissue damage through digestion of structural compounds and substrate tissues. They also increase permeability of the vessels and produce inflammatory mediators such as histamine, which allow more bacteria to enter into the bloodstream and increase septicemia. 85 Ulilysin is also a metalloproteinase from the pappalysin family, produced by archaea bacterium Methanosarcina acetivorans. 86 In the past, metalloproteinases such as CpaA have been identified in A. baumannii, which have domains similar to pappalysins and affect human coagulation disorders, 87 but there has been no report of ulilysin-like metalloproteinase production by this bacteria.
The results also showed that HP7 was very similar to oxidoreductase enzymes from the short-chain dehydrogenases/reductases (SDRs) family. This oxidoreductase protein interacts with a protein called DrgaA and a transcription-regulating protein of the TetR family. DrgaA is a protein that has the binding domains to FMN, FAD, or NAD(P)H, and nitroductase activity. The activity of this enzyme in some pathogenic bacteria has been shown to sensitize them to nitro drugs such as nitrofuran.88,89 The members of TetR family are also one of the important regulators for the transcription of genes involved in antibiotic resistance, drug efflux, metabolism, and quorum sensing. 90 SDRs are a very large group of NAD- and NADP-dependent oxidoreductase enzymes that have two domains of NAD and substrate binding. Proteins with 3 beta-hydroxy-5-one steroid dehydrogenase and uridine diphosphate-glucose-4-epyrerase are also part of this family. 91 In this regard, alcohol dehydrogenase has a limited substrate range and alcohol dehydrogenase of the bacterium Sphingobium yanoikuyae, which has a very similar sequence with HP7, uses ketones that have their carbonyl groups attached to two hydrophobic groups as substrates. 92 As shown in Supplementary Table S3, this protein has a KR domain, most commonly found in polyketide and fatty acid synthase enzyme, which catalyzes the first step in the reduction of beta-carbonyl nuclei in the growing chain of polyketide. These enzymes use NADPH to reduce the keto group to hydroxy. As shown in Supplementary Table S3, in the same region, the structure of SCOP d1hu4a has been also identified, which is related to carbonyl reductase/20beta-hydroxysteroid dehydrogenase protein.
HP7 with E-value = 21e-1.2577 and 30% identity is similar to mutated 3-oxoacyl-acyl carrier protein reductase (fabG). FabG is a protein involved in the lipid metabolism and fatty acid biosynthesis. Triclosan biocide stops the final reduction of fatty acids in their biosynthesis pathway. Pathogenic bacteria have also become resistant to this biocide by mutating its target. 93 These results have also confirmed involvement of this HP in the metabolism of fatty acids.
HP8 is similar to BapA prefix-like domain containing and adhesin proteins. Its similarity to nonfimbrial adhesin in Salmonella enterica, called SiiE, was also predicted. The results in Table 4 also showed its extracellular relationship with type I secretory system (T1SS). The presence of an FN3 domain in this protein is demonstrated in Supplementary Table S3. T1SS in Gram-negative bacteria is involved in the secretion of pathogenic factors, such as adhesines. 94 Salmonella are also equipped with numerous adhesive structures such as BapA and SiiE, which are secreted by T1SS.95,96 Moreover, fibronectin is a dimer glycoprotein connected to its subunits by disulfide bridges. This molecule is involved in cellular connectivity and consists of known domains seen in animal proteins, 97 but its presence in bacteria has been also observed. 98 The presence of this domain in the proteins of the Bap and SiiE family was proven. A. baumannii also has a large protein called Bap that plays a role in the formation of biofilms and its attachment. 99 Similar to this protein with different lengths and sequences, some have been detected in different strains of A. baumannii. 100 It seems that this HP is also an important common attachment mediator protein among A. baumannii strains. The classification of this protein as a pathogenic agent by VICMpred (Table 6) reinforces this hypothesis.
The results of Supplementary Table S6 predicted HP11 as urea carboxylase-associated protein with E-value of 1e-164. This protein was identified to interact with the urea carboxylase, urea carboxylase-associated protein 2, allophanate hydrolase, glutamate synthase, 5-methyltetrahydrofolate-homocysteine methyltransferase, ABC transporter/permease protein, and a protein from the TIGR00370 family. Urea carboxylase is also known as an enzyme that catalyzes a biotin-dependent and ATP-dependent two-step chemical reaction. The given enzyme consists of biotin carboxylase (BC), carboxyltransferase (CT), and biotin carboxyl carrier (BCCP) domains. The activity of this enzyme also results in the production of urea-1 carboxylate (allophanate). 101 Alfonate hydrolase converts alfonate into ammonium and carbon dioxide. Thus, this enzyme is required to use urea as a source of nitrogen. 102 The enzyme 5-methyltetrahydrofolate-homocysteine methyltransferase, also called methionine synthase, converts homocysteine into methionine to be involved in the production of amino acids. This enzyme is a urea-resolved apoenzyme separated from halo-enzyme by urea. 103 Accordingly, urea is an important nitrogen source for many microorganisms, but its transmitters are only found in limited bacteria, which transfer urine from the outside into the cell in the absence of sources of nitrogen. ABC transporter/permease enzymes have been also shown to transfer urea in cyanobacteria, 104 and it seems that A. baumannii also uses this type of transporter. In general, evidence suggests that HP11 is a protein involved in the urea catalysis used for the synthesis of amino acids.
The results presented in Fig. 1 showed that this HP with a degree of 8, eccentricity value of 1, and betweenness centrality of 0.18 plays a very important role in this process. The importance of urea metabolism in A. baumannii was further identified as a result of a study in 2016, in which it was determined that the given bacteria should be adapted to limited nutritional metals in the host body. An intrinsic immune system protein, called calprotectin, chelates metals in the body, but A. baumannii survives within the body under these conditions by use of urea as the sole source of nitrogen. Furthermore, the urea carboxylase gene is located in an operon encoding a transporter from natural resistance-associated macrophage protein (NRAMP) family, which uses a hydrogen concentration gradient for transfer and accumulation of Mn ions. Thus, A. baumannii can survive in the absence of metals in the body. 105
HP15 is predicted to be a member of PhoX phosphatase family (Table 5) and similar to PhoX alkaline phosphatase from Pseudomonas fluorescens (Supplementary Table S2). The results of Supplementary Table S6 demonstrated its association with the Tat secretion pathway. The genes present in Pho regulon are involved in the sensation of amount of inorganic phosphate in the environment. The expression of the genes of this regulon are also correlated with lack of phosphorus in the environment. PhoX is one of these genes whose product is an alkaline phosphatase. 106 It should be noted that alkaline phosphatases are phosphomonoesterases that detach the phosphate from phosphate-containing organic sources. 107 The release of this enzyme through the Tat pathway has been introduced in some bacteria. 108 It has been also introduced as a pathogenic factor that is, Pseudomonas aeruginosa,109,110 which is consistent with the results of the VICMpred software (Table 6). The prediction of this function is also in agreement with the prediction of this protein as an extracellular protein using the nonclassical secretion pathway (Table 4).
Immunological analyses
Proteins that are potential candidates for vaccine development should be exposed to extracellular spaces to be available to the host immune system. 111 Therefore, four HPs, HP4, HP6, HP8 and HP15, were selected among functionally important HPs for immunological predictions. Since inhibiting the functions of normal bacterial flora results in adverse side effects and in colonization of the gut by pathogens, 112 a comparative similarity analysis of vaccine candidate HPs with proteins of human gut microbiota was performed by BLASTp. Results indicated that there was no protein homolog among gut flora proteome. As seen in Table 7, upon VaxiJen prediction, all four investigated HPs were antigens. HP4 and HP6 were allergens and HP8 and HP15 were potential allergens according Algpred prediction results. Referring to predictive results of antigenic regions (Supplementary Table S7), the number and sequence of B cell (Supplementary Table S8) and T cell epitopes (Supplementary Tables S9 and S10) and IC50 of common epitopes predicted by PropredI and Propred (Table 8), and summarized results of used softwares for prediction of immunological properties (Table 7), the following information was obtained.
The Summarized Results of Used Softwares for Prediction of Immunological Properties of Four Hypothetical Proteins HP4, HP6, HP8, and HP15
Location, Sequence, and Half Maximal (50%) Inhibitory Concentration of Common Epitopes Predicted by PropredI and Propred
IC50, half maximal (50%) inhibitory concentration.
HP4 has 11 antigenic regions, 4 B cell epitope, 46 MHC I-binding epitopes, and 34 MHC II-binding epitopes. This protein has an epitope with the sequence of “IRVITTAFL,” which is capable of binding to 25 types of MHC II alleles. The ability of epitopes to bind to multiple HLA alleles is a positive factor in the development of a universal epitope-based vaccines. 113 HP6 has 12 antigenic regions, 6 B-lymphocyte-binding epitopes, 48 MHCI-binding epitopes, and 44 MHCII-binding epitopes. The eptipope with the sequence of “FRNNGKPFI” is predicted to bind 30 types of MHC II alleles. Moreover, there was an epitope with the sequence of “ILLLISGTL,” which binds both classes of MHCs (three MHCI alleles and one MHCII allele). The IC50 value of this common epitope was 79.07. Epitopes with IC50 <100 nM are usually classified as a good binder. 114 Based on prediction results, HP8 possessed 29 antigenic regions, 23 B-cell epitopes, 89 MHC I-binding epitopes, and 66 MHC II-binding epitopes. Epitopes with sequences of “RGLNHVTDL” and “FSYKFTPPL” could bind to 10 types of MHC I alleles. This protein contained eight common epitopes predicted by both PropredI and Propred, six of which showed IC50 < 100 nm. Finally, HP15 included 27 antigenic determinants, 28 B-cell epitopes, 100 MHC I-binding epitopes, and 63 MHC II-binding epitopes. Interestingly, there was an epitope with sequence of “LRRFLVGPK” in the list of Propred-predicted epitopes that could bind to 32 types of MHC II alleles. Three common MHC I and MHC II binding epitopes were predicted in this protein. “YLTTEENFI” is one among them, which has very low IC50 of 1.12. To our knowledge, there is no report about in silico investigation of common HPs of antibiotic-resistant A. baumannii as drug or vaccine candidates using methodology of this article. Moreover, the HPs suggested using different methodologies of reverse vaccinology were not similar to those reported in this study.17,115
Conclusion
The death/year caused by antimicrobial resistance will be equal to present cancer-caused mortality after 2050. Due to antibiotic resistance, A. baumannii is considered one of “priority pathogens” in World Health Organization (WHO) list. Today, many efforts are being made to generate therapeutic methods that cover elimination of all possible resistant strains. In this study, functional annotation of common 18 HPs from hypervirulent and antibiotic-resistant strains of A. baumannii was carried out using various in silico approaches and functions assigned to them. In this study, the prediction results showed possible functional importance of the seven HPs in the successful pathogenicity of the pathogen in the host. Moreover, immunological analyses with prediction softwares indicated four HPs to be probable vaccine candidates. However, in vitro and in vivo tests should be performed to prove the suitability of these HPs for drug and vaccine development. There are significant number of uncharacterized genes and their products known as HPs. In the current antibiotic and drug resistance era, annotation of these HPs could be beneficial to find new treatment and diagnosis strategies of disease and vaccine candidate introduction. However, this field still needs advanced researches using more innovative software and specialized databases to characterize the predicted proteins more precisely, in the future.
Footnotes
Acknowledgment
The authors appreciate the University of Tabriz for supporting this work.
Disclosure Statement
The authors declared no conflict of interests.
Funding Information
This research was financially supported by a research grant (Number 2.112376.52) for an MSc thesis from University of Tabriz, Iran.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.