
Abstract
Beyond the emergence of plasmid-encoded mechanisms, mutation within the pmrAB genes remains one of the primary colistin resistance mechanisms in Escherichia coli. However, the mechanisms of high-level colistin resistance (HLCR) have not been elucidated. In this study, we evaluated the HLCR mechanisms in five colistin-susceptible Avian pathogenic Escherichia coli (APEC) isolates after colistin exposure. Three PmrB substitutions (G19R, L167P, V88E) and two PmrB sequence duplication (PmrB-sd) mutations (68-77dup and 94-156dup) were detected. Chromosomal replacement and deletion mutagenesis revealed the two PmrB-sd mutations contribute to, but are not fully responsible for, HLCR in APEC strains. Quantitative reverse transcription/polymerase chain reaction (qRT-PCR) revealed that the PmrB-sd induction mutants showed an increased pmrAB transcript level and the PmrB-sd reversion mutants exhibited a reduction of pmrAB expression. All five induction mutants exhibited decreased minimum inhibitory concentrations to florfenicol and tetracycline. In addition, four mutants (G19R, L167P, V88E, and 94-156dup) and two mutants (68-77dup and 94-156dup) also displayed increased sensitivity to ceftiofur and gentamicin, respectively. Zeta potential measurement of the induction mutants showed that there was less negative charge on the cell surface compared with its parental strains in the absence of colistin. The induction mutants also showed an increase of lag time and decrease of fitness. In summary, the identification of novel PmrB-sd mutations contributing to HLCR is helpful to broaden the knowledge of colistin resistance. Attention should be paid to the use of colistin for the treatment of infections caused by APEC strains.
Introduction
Avian pathogenic Escherichia coli (APEC) causes a variety of diseases, principally the respiratory and systemic infections, and is responsible for great economic loss to poultry worldwide.1,2 Subtractive hybridization and phylogenetic studies have suggested that APEC strains may act as zoonotic pathogens and reservoir of virulence causing human infections.2–5 As a subgroup of extraintestinal pathogenic E. coli, APEC strains have emerged as a large reservoir to critically important antimicrobials in the past decade, especially fluoroquinolones and cephalosporins.6,7 Moreover, increasing resistance to colistin has been reported in APEC in recent years, which is of worldwide concern.8–11
Colistin is a polypeptide antibiotic that has been reintroduced as one of the last-resort antibiotics in response to the increasing incidence of multidrug-resistant Gram-negative infections, including E. coli.12,13 However, acquired colistin resistance mediated by plasmids or chromosomal mutations has been reported. Since the first report of plasmid-mediated colistin resistance gene mcr-1 in 2015, the mcr-1 gene and its variants (mcr-2 to mcr-9), which encode phosphoethanolamine transferases modifying the lipid A, have been isolated from livestock, food, and humans all over the world.14–22 The mcr genes normally confer low-level colistin resistance (2–8 mg/L).15,23
Mutations in genes involved in modification of the lipopolysaccharide (LPS), primarily the two-component systems, pmrAB and phoPQ, are responsible for the chromosomal colistin resistance in E. coli.23–25 They usually display low- to moderate-level colistin resistance (<32 mg/L).23–25 The activation of PmrAB leads to upregulation of genes responsible for synthesis of cationic groups (L-Ara4N and pEtN) and/or their addition to the LPS. 23 Mutations within the pmrAB genes have been well documented as being responsible for colistin resistance through activation of PmrAB in many bacterial species, including Klebsiella pneumoniae, Salmonella enterica, Pseudomonas aeruginosa, and Acinetobacter baumannii.26–30
Recently, a study indicates that the acquisition of mcr-1 can facilitate the selection of high-level colistin-resistant (minimum inhibitory concentration [MIC] ≥32 mg/L) mutations in PmrAB of E. coli. 31 However, the underlying mechanisms involved in HLCR remain to be discovered. In this study, we identified HLCR mutations from colistin-susceptible APEC isolates after colistin exposure and revealed that two novel mutations of sequence duplication within pmrB gene (pmrB-sd) contribute to HLCR by using comprehensive approaches.
Methods
Bacterial strains and antimicrobial susceptibility testing
Colistin-susceptible APEC strains used in this study were previously isolated from diseased ducks in China, 2012 (Table 1). Isolates were identified by polymerase chain reaction (PCR) using primers targeting 16S rRNA gene as previously described.32,33 All E. coli strains were cultured on Luria Broth (LB) broth or agar at 37°C and stored in glycerol at −80°C until use.
Avian Pathogenic Escherichia coli Strains and Their Mutant Pairs Evaluated in This Study

The MICs of colistin, ceftiofur, gentamicin, enrofloxacin, florfenicol and tetracycline were determined using the broth dilution method as recommended by the CLSI guidelines. 34 E. coli strain ATCC 25922 was used as the control strain. All tests were performed independently at least three times.
In vitro selection of colistin-resistant induction mutants
Spontaneous induction assay was performed as previously described. 15 In brief, the clinical isolates were recovered on LB plates and colonies of each strains were grown in LB broth. The bacterial cultures were incubated on LB plates supplemented with increasing concentrations of colistin (from 1/2 × MIC to 128 μg/mL). Colonies that exhibited highest colistin MIC were passaged on colistin plates every day for 10 days (∼1,000 generations) or until the highest MIC to colistin was reached. Colonies of each strain that reached to highest colistin MIC were stored for further investigation.
PCR and nucleotide sequencing
Genes of pmrAB, phoPQ, mgrB, and lpxACD, which is reported to be involved in colistin resistance in E. coli, were amplified with primers in both parental strains and induction mutants (Supplementary Table S1). PCR amplicons were purified and sequenced to detect the possible mutants in these genes of the isolates prior and after colistin induction assay.
Reconstruction of pmrB/pmrB-sd replacement and pmrA deletion mutants
Two colistin-resistant induction mutants with PmrB-sd, IND-91 (PmrB68–77dup), and IND-107 (PmrB94–156dup), were replaced with wild-type PmrB, designated IND-91-REV, and IND-107-REV, by using λ-Red homologous recombination system with primers listed in Supplementary Table S1. 35 All reconstructed strains were confirmed with PCR followed by sanger sequencing. For each constructed mutant, a corresponding strain was constructed as control only carrying an FRT-scar.
Furthermore, the parental strain APEC-91 was replaced with PmrB68-77dup from strain IND-91, designated pmrB68-77dup. A pmrA insertion/deletion mutant by homologous recombination with terminal pmrA sequences and a chloramphenicol resistance cassette, designated ΔpmrA, was also reconstructed into PmrB68-77dup mutant IND-91.
Quantitative reverse transcription/polymerase chain reaction
The mRNA expression levels of pmrA and pmrB genes were detected by qRT-PCR. Gene dnaE was used as internal control. Total RNA of each strains was extracted from exponentially growing culture (OD600 = 0.5) using the RNAiso Plus (TaKaRa Bio, China) following the manufacturer's instructions. RNA was used as the template for cDNA synthesis using the PrimeScrip RT Reagent Kit with gDNA Eraser (TaKaRa Bio). The cDNA products of each sample were amplified using primers designed by using software Oligo7.0 (Supplementary Table S1). All reactions were performed in triplicate and the experimental data were analyzed using the 2−ΔΔCT method.
Zeta potential measurement
Zeta potential measurements were performed as previously described with minor modifications. 36 Briefly, bacterial cells were harvested from broth culture and washed twice with Milli-Q™ water. Washed cells were resuspended in the test solution to prepare bacterial suspensions containing ∼1 × 108 cfu/mL. The electrophoretic mobility of bacterial cells was measured with a zeta potential analyzer (Brookhaven, NY) in triplicate.
Transmission electron microscope analysis
The parental strains and induction mutants treated without or with 4 mg/L colistin were examined with transmission electron microscope (TEM) analysis as previously reported. 37 In brief, bacterial cells were grown to reach an optical density of 0.5 and harvested from 1 mL of suspension by centrifugation at 12,000 rpm for 10 minutes. The cell pellets were resuspended in 4% glutaraldehyde and incubated for 4 hours at room temperature. The postfixation was carried out with 2% glutaraldehyde for 1 hour, followed by dehydration through a graded ethanol series. The cell pellets were washed and dissolved in tert-butyl alcohol and sent for TEM analysis.
Bacterial growth curve and competition assay
All strains were grown in LB broth overnight, and diluted to an OD600 of 0.02 in LB broth for 24 hours at 37°C with shaking at 180 rpm. OD600 values were measured every hour for successive 24 hours.
Culture of each competitor was adjusted to OD600 = 0.2, mixed in 1:1 ratio, and diluted 1:100 in 5 mL daily for 10 days. The cfu of the competitors was counted by plating onto medium with or without colistin. The relative fitness (w) was determined in competition experiments in 10 replicates and repeated at least twice, as previously described. 38
Ethics statement
The study does not involve animal/human experiments. Ethics statement is not applicable.
Results
Mutations that confer HLCR in induced mutants
To elucidate the detailed mechanisms of induced resistance to colistin, we selected five colistin-susceptible APEC isolates for colistin induction in the presence of colistin. Five HLCR mutants with colistin MIC equal or more than 128 mg/L were derived from parental strains (Table 1). Sequencing analysis showed that these laboratory-derived resistant strains contained no mutations in pmrA, phoPQ, mgrB, or lpxACD, however, mutations in pmrB gene existed. Three mutants, IND-9, IND-36, and IND-53 had point mutations in pmrB resulting in amino acid substitutions of G19R, L167P, and V88E, respectively (Fig. 1 and Table 1). Of note, other two mutants were identified with novel sequence duplication within pmrB (PmrB-sd) from nucleotide 202 to 231 in strain IND-91 with 10 amino acids longer (68SLIVPGVFMV77, PmrB68-77dup) and from nucleotide 280 to 468 in strain IND-107 with 63 amino acids longer (94PLAELQKELEARTADNLTPIAIHSATLEIEAVVSALNDLVSRLTSTLDNERLFTADVAHELRT156, PmrB94-156dup) (Table 1). Figure 1 and Supplementary Table S2 shows the domains of PmrA/PmrB and positions of induced mutations conferring colistin resistance in this study as well as previously published mutations.24,25,31,39,40 After subculturing in the absence of colistin, the PmrB-sd mutants IND-91 and IND-107 showed no reduction of colistin MIC, suggesting that the acquisition of this resistance phenotype was stable.

Contribution of PmrB-sd mutations to HLCR
To confirm the role of PmrB-sd mutations in HLCR, PmrB revertants by replacing PmrB68-77dup and PmrB94-156dup with wild-type PmrB in IND-91 and IND-107 were reconstructed, respectively. The results showed that the colistin MIC value decreased to a level comparable to those of parental strains APEC-91 and APEC-107 (Table 1). When the wild-type PmrB was replaced with PmrB68-77dup in parental strain APEC-91, designated pmrB68-77dup, a 16-fold elevated colistin MIC (16 mg/L) was observed, although lower than that in colistin-induced mutant IND-91 (128 mg/L). The pmrA deletion mutant exhibited decreased colistin MIC values (1 mg/L) comparable to that of its parental isolate IND-91 (1 mg/L). Altogether, these results revealed that PmrB-sd mutation contributed to, but were not fully responsible for, HLCR in APEC strains, and PmrA was required for PmrB-sd mutation-mediated HLCR in APEC strains.
pmrAB mRNA expression level of HLCR mutants
The mRNA expression of pmrAB genes in pmrB-sd mutants was compared with expression in parental isolates by qRT-PCR. In IND-91, the transcription of pmrA and pmrB was upregulated 2.5- and 4.1-fold, respectively, compared with APEC-91 (Fig. 2). Similarly, the pmrA and pmrB transcript levels in IND-107 was also upregulated 2.0- and 2.4-fold compared with its parental strain, respectively (Fig. 2). Correspondingly, the pmrA and pmrB transcript levels in strains IND-91-REV and IND-107-REV was reduced to a level similar to those in wild-type strains (Fig. 2).
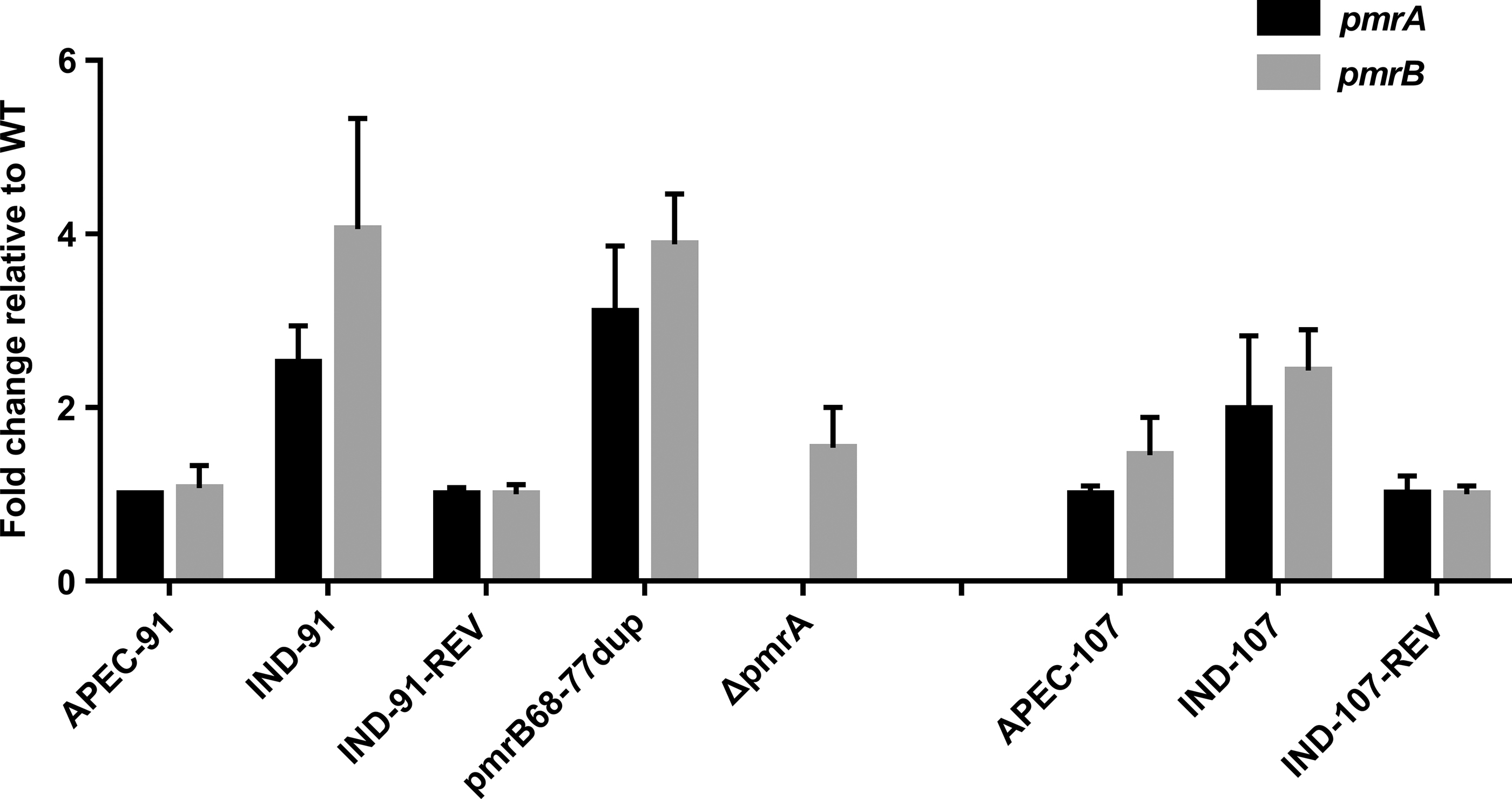
The transcript level of pmrAB in HLCR mutants compared with the wild-type strain. All reactions were performed in triplicate and the fold change in transcription were calculated using the 2−ΔΔCT method. HLCR, high-level colistin resistance.
Increasing susceptibility to other antimicrobials of HLCR mutants
To investigate the antimicrobial susceptibility profiles to other antimicrobials after colistin induction, MICs of ceftiofur, gentamicin, enrofloxacin, florfenicol, and tetracycline against the five HLCR mutants were measured. As shown in Table 1, all five mutants had decreased MICs to florfenicol and tetracycline. In addition, four mutants (G19R, L167P, V88E, and 94-156dup) and two mutants (68-77dup and 94-156dup) also displayed increased sensitivity to ceftiofur and gentamicin, respectively. No change of MICs for enrofloxacin was observed in both enrofloxacin-resistant and enrofloxacin-susceptive strains (Table 1).
Zeta potential and cell wall morphological differences of HLCR mutants
The membrane zeta potential was observed in colistin-susceptible parental isolates and their derived colistin-resistant induction mutants. In absence of colistin, zeta potential values of the parental isolates were ranging from −55.2 ± 0.945 to −36.2 ± 0.451 mV, while all colistin-resistant mutants showed less surface charge than their parental strains (Fig. 3). The magnitude of zeta potential changes varied according to the individuals. The largest increased level of mean zeta potential was observed in strain IND-36, from −51.9 to −28.4 mV, when compared with its parental APEC-36. Strain IND-107 exhibited the smallest increase level of surface charge (2.5 mV) compared with its parental APEC-107 (Fig. 3). In the presence of colistin, all pairs of APEC strains showed a similar magnitude of surface charge neutralizing effect, ranging from −13.0 ± 0.252 to −3.86 ± 0.564 mV (Fig. 3).

Zeta potential (mean + standard deviation) of APEC parental strains and corresponding induction mutants in the absence and presence of colistin. APEC, Avian pathogenic Escherichia coli.
The morphological differences between colistin-susceptible parental isolates and derived colistin-resistant mutants were evaluated by TEM. In the presence of colistin (2 mg/L), the colistin-susceptible parental isolates presented a discontinuous cell wall, markedly diffuse outer membrane, or extensive fibrils (Fig. 4A–E), whereas the mutants were uniform in structure with well-preserved cell walls (Fig. 4A1–E1). In the absence of colistin, all the strains exhibited an even cytoplasm and intact cell wall. The TEM macrographs of one strain pair, APEC-91 and IND-91 as examples, are shown in Fig. 4F and F1.

Representative TEM macrographs of APEC parental isolates cultured in the presence of
Bacterial growth and fitness cost of HLCR mutants
Growth curves were measured for the colistin-susceptible parental isolates and their derived colistin-resistant induction mutants. Except for IND-53, growth of all mutants to 0.4 of OD600 were decelerated approximately 1 to 3 hours compared with those of their parental strains (Fig. 5). However, no significant differences in the growth rate were observed during the late logarithmic phase.

In vitro growth of APEC parental isolates and the derived colistin-resistant mutants.
The fitness cost (∼1,000 generations) of derived colistin-resistant mutants was determined by competition assay. As expected, all of the induction mutants exhibited less fitness compared with parental strains (Fig. 6). The fitness index of each pair ranged from 0.61 ± 0.060 (strain IND-36) to 0.67 ± 0.12 (strain IND-91). The results indicate that the PmrB mutations conferring HLCR led to a fitness decrease.

Relative competitive fitness of all five induction mutants compared with its parental strains. The relative fitness (w) of ∼1,000 generations was determined in competition experiments for 10 days in 10 replicates and repeated at least twice.
Discussion
Colistin has been extensively used in veterinary medicine as feed additives since 1960s, mainly for the prevention and treatment of Enterobacteriaceae infections. 10 However, colistin resistance mediated by acquisition from plasmids or chromosomal mutations has been increasingly reported. Recent large epidemiological study showed that 72.6% of the colistin-resistant E. coli isolates harbored convergent mutations in the PmrAB proteins, highlighting the role of this two-component system in colistin resistance in E. coli. 41 Although some polymorphism in the pmrAB genes of colistin-resistant E. coli has been reported, the involvement of these mutations in the colistin resistance has not formally been demonstrated.24,25,40,42 In addition, a very recent report showed that pmrAB mutations contribute to the selection of HLCR in E. coli. 31 Currently, HLCR mechanisms have been documented involving mutations in crrB gene in K. pneumoniae,43,44 in colRS and cprS genes in P. aeruginosa, 45 and in lpxACD genes or inactivation of H-NS regulator in A. baumannii,46,47 but not reported in E. coli. In this study, we identified three PmrB substitutions and two PmrB-sd mutations in laboratory-derived strains contributing to HLCR. Further differences were observed in the pmrAB mRNA expression, zeta potential, cell wall changes, growth, and fitness when comparing the mutants with their parental strains.
The three PmrB substitutions and two PmrB-sd mutations reported in this study are novel in E. coli. One of the substitutions, PmrB-L167P, has been previously reported to be involved in colistin resistance in P. aeruginosa. 48 Sequence duplications within PmrB have been described within the transmembrane domain 1 (TM1) of PmrB of both clinical (PmrB15-24dup) and laboratory-derived (PmrB17-26dup) A. baumannii isolates, leading to a longer protein by addition of 10 amino acids.49,50 In this study, PmrB68-77dup mutations occurred within TM2 of PmrB resulting in 10 amino acids longer, and PmrB94-156dup within intramembrane domain of HAMP and HisKA in PmrB leading to an increase of 63 amino acids (Fig. 1). We speculated that the PmrB substitution and PmrB-sd mutations may activate the PmrB sensing, resulting in phosphorylated activation of the PmrA regulator, and therefore increased the transcription level of the pmrCAB operon, as confirmed by our qRT-PCR analysis (Fig. 2). This finding is consistent with previous studies.24,40 Previously identified PmrB mutations normally confer low- to moderate-level colistin resistance (<32 mg/L),23–25 which cannot explain the HLCR in our study, therefore, we evaluated the contribution of the novel PmrB-sd mutations in HLCR by replacing of PmrB-sd with wild-type PmrB in induction mutants as revealed by reversion of the colistin MIC and pmrAB expression to the original level (Table 1 and Fig. 2). But when replaced, the wild-type PmrB with PmrB-sd in wild-type strain, the PmrB-sd parental strain pmrB68-77dup exhibited moderate-level colistin resistance (16 mg/L). Identified mutations in pmrA, phoPQ, mgrB, and lpxACD reported in other Gram-negative bacteria were not found in this study.23,46,51,52 The above results indicate that PmrB-sd mutations contributed to, but were not fully responsible for, HLCR in APEC strains. Two possible mechanisms might be contributed to HLCR in E. coli: (1) pmrB mutation coupled with mutation in genes involved in LPS modification and hyperproduction of capsular polysaccharide, or encoding porins and efflux pumps23,53–55; (2) novel undiscovered mechanisms confer HLCR. However, the underlying mechanisms remain to be further investigated.
It has been documented that the colistin-resistant isolates display increased sensitivity to nonpolymyxin antibiotics in A. baumannii.47,56 To determine the impact of PmrB mutations in E. coli, we detected the MICs of five classes of clinically relevant antimicrobials for the parental strains and mutants (Table 1). All five mutants with PmrB mutation exhibited decreased MICs to florfenicol and tetracycline, with four mutants with PmrB mutation (G19R, L167P, V88E, and 94-156dup) and two mutants with PmrB mutation (68-77dup and 94-156dup) also displayed increased sensitivity to ceftiofur and gentamicin, respectively. The increased sensitivity of the mutants with PmrB mutation to nonpolymyxin antimicrobials may be resulted by modification of LPS, with altered outer membrane and increased permeability to antimicrobials that normally do not penetrate through the outer membrane.47,56–58
Zeta potential differences between each mutant with PmrB mutation and parental strain pairs were measured to address outer membrane changes as reflected by decreased cell surface negative charge. After colistin exposure, all pairs of APEC strains showed similar magnitude of surface charge neutralizing effect (Fig. 3), and as expected, the colistin-susceptible parental isolates presented a discontinuous cell wall, whereas the mutants presented a well-preserved cell wall. In the absence of colistin, although both the parental strains and mutants presented a well-preserved cell wall (Fig. 4F and F1), the mutants showed less surface charge than their parental strains (Fig. 3). This difference in surface charge might be dependent on the modification of the cell surface LPS resulting from mutations in pmrB, and potentially other undiscovered mechanisms. 36
All the mutants with PmrB mutation showed an obvious increase of lag time when compared with its parental isolates, suggesting the presence of fitness cost in these mutants. A competition assay between mutants with PmrB mutation and parental strains revealed a reduction of fitness in mutants with PmrB mutation. The results are consistent with previous studies in E. coli and other Gram-negative bacteria.24,59,60 The fitness cost caused by chromosomal mutations may explain the lower percentage of colistin-resistant isolates in these years until the emergence of plasmid-mediated colistin resistance. 58 However, Bourrel et al. have recently reported that extremely high rate of the colistin-resistant E. coli isolates are mediated by PmrAB mutations. 41 This may due to the accumulation of compensatory mutations in response to the emergence of colistin-resistant virulent strains with fitness cost after prolonged colistin administration.41,61
In conclusion, two novel sequence duplication mutations within pmrB gene contributing to HLCR were identified in this study, which is helpful to broaden the knowledge of colistin resistance caused by sequence duplications. Attention should be paid to the use of colistin for the treatment of infections caused by APEC strains. A long-term verification of bacterial fitness and evaluation of virulence in HLCR mutants should be addressed in future studies.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Key R&D Program of China (2018YFD0500300), the National Natural Science Foundation of China (31572567 and 31702292), the Natural Science Foundation of Jiangsu Province (BK20170710), the China Postdoctoral Science Foundation (2017M611841 and 2018T110515), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.