
Abstract
A new member of the class metallo-β-lactamase (MBL), New Delhi metallo-beta-lactamase 1 (NDM-1) has emerged recently as a leading threat to the treatment of infections that have spread in all major Gram-negative pathogens. The enzyme inactivates antibiotics of the carbapenem family, which are a mainstay for the treatment of antibiotic-resistant bacterial infections. This review provides information about NDM-1 spatial structure, potential features of the active site, and its mechanism of action. It also enlists the inhibitors/compounds/drugs against NDM-1 in various development phases. Understanding their mode of inhibition and the structure-activity relationship would be beneficial for development, synthesis, and even increasing biological efficacy of inhibitors, making them more promising drug candidates.
Introduction
A
Many resistance mechanisms are known so far, with the familiar being the production of enzymes that degrade antibiotics, for example, beta-lactamases, 3 which hydrolyze the lactam ring of the drug and lead to its inactivation. 4
Ambler classification categorizes β-lactamases into four distinct classes (A through D). Classes A, C, and D, as serine-β-lactamases (SBLs), work through serine hydrolyses whereas class B metallo-β-lactamases (MBLs) require one or two zinc ions in the active site to mediate hydrolysis. 5 MBLs class B enzymes are further divided into B1, B2, and B3 subclasses, of which B1 class has emerged as the most clinically significant enzyme. 6 to be added. Besides, the location of MBLs on highly transmissible plasmids, 7 bacteria equipped with MBLs, has shown a broad spectrum of activity against almost all β-lactam antibiotics such as penicillin, cephalosporin, aztreonam, and even carbapenems by degrading them. These antibiotics are considered to be the last line of defense. Moreover, MBLs are insensitive to clinically available inhibitors.8,9 Thus, MBLs are considered to be more dangerous to human beings by increasing public health risk.
A new member of MBLs is the New Delhi metallo-β-lactamase 1 (NDM-1) that has rapidly emerged as a leading threat to the treatment of bacterial infections. 7 The NDM-1 encoded by the blaNDM-1 gene was initially identified in a Klebsiella pneumoniae isolate in 2008. 7 Since then, NDM-1 has been found in at least 180 variants in different strains, including Escherichia coli, A. baumannii, Vibrio cholera, Stenotrophomonas maltophilia, and Pseudomonas putida. Very few antibiotics are available for the treatment of patients infected by the bacteria carrying NDM-1.10–15 Understanding the mechanism of antibiotic degradation by NDM-1 protein and the potential role of its active site would be helpful in discovering or redesigning new β-lactam antibiotics for the NDM-1-producing bacterial strains. Screening for new inhibitors alone or partnering with known β-lactam antibiotics against NDM-1 activity can work appropriately by interrupting the formation of a bacterial cell wall, which is regarded as the quick and efficient route of bacterial cell death. Looking for potent and safe inhibitors to ensure the activity of antibiotics against NDM-1-producing bacterial strains is, thus, an urgent requirement.
Structure and Active Site of NDM-1
With dimensions of 50 × 40 × 40 Å, the NDM-1 enzyme is a compact globular structure that displays typical conical αββα sandwich architecture of the MBL superfamily. The enzyme has two central antiparallel β sheets flanked by two pairs of α-helix. The left portion of the NDM-1 molecule has two α-helix (α1 and α2) and seven antiparallel β-strands (β1–β7), whereas the right subdomain consists of the remaining two α-helices (α3 and α4) and four antiparallel β-strands (β8–β11). 16
The active site of NDM-1 enzyme (Fig. 1A) follows a two-ion catalytic mechanism, a characteristic of the MBL superfamily. The NDM-1 active site centered by two zinc ions (Zn I and Zn II) is located at the external edge of a ββ sandwich surrounded by His120, His122, Asp124, His189, Cys208, and His250. Tetrahedral and trigonal pyramidal coordination spheres are displayed by Zn I and Zn II, respectively. His120, His122, His189, and one hydroxyl of Asp124 are referred to as histidine sites in MBL superfamily forms, and they display tetrahedral coordination; whereas Asp124-Cys208-His250 referred to as a cysteine site displays a trigonal pyramidal coordination sphere. 5 The two zinc ions contact each other with the distance of 3.2 Å bridged by the side chain of Asp124.17,18 The active site of NDM-1 consists of a deep cavity formed by the loop regions between β5-α2 and β10-α4, with deeply buried catalytic zinc ions. This cavity is clamped by the loop regions between Thr119-Met126 and Ser217-Asp225 referred as “ceiling” and “floor.” Loop (Met67-Gly71) directs the opening and closing of the active site (Fig. 2B) for product release and substrate binding via conformational changes and, thus, acts as a doorkeeper for the NDM-1 active site. This “ceiling” and “floor” are largely conserved in B1 and B2 subclasses of MBL. However, the doorkeeper loop of NDM-1 has less similarity with known MBL structures. 16
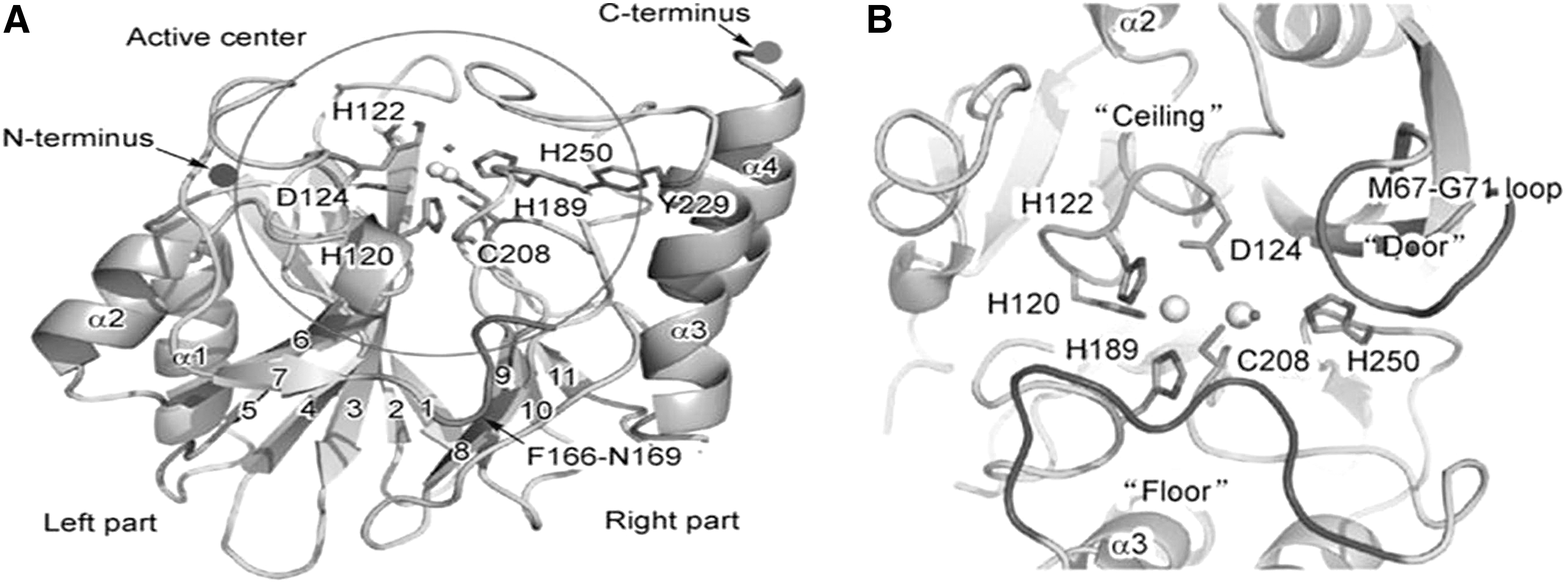
Crystal structure of overall ribbon diagram of New Delhi metallo-β-lactamase:
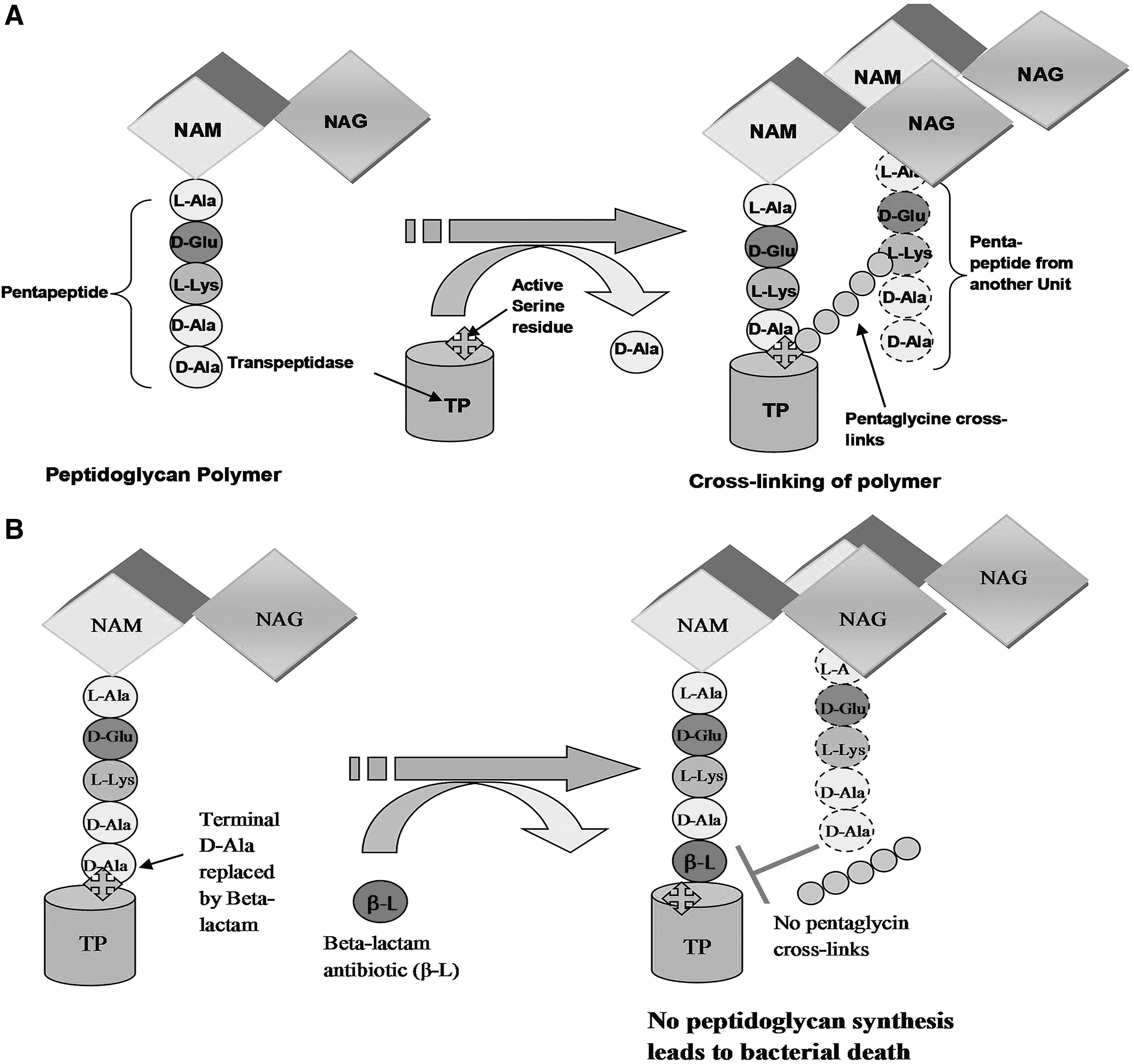
Schematic representation of peptidoglycan cross-linking by bacterial transpeptidase enzyme (TP).
A potential feature of NDM-1 active site
The N-terminus loop region of subclass B1comprising residues 61–6517 is absent in subclasses B2 and B3 of the MBLs superfamily. 5 A tunnel-shaped cavity is formed at the groove of the active site by the interaction of the N-terminus loop with hydrophobic side chains of the substrate or inhibitor. However, this interaction results in the blockage of the active site. 5 Deletion of loop results in weakening of substrate binding by the enzyme and, thus, seriously affects the enzyme activity except for imipenem. The loop region that covers the active site features great mobility during substrate binding and shows different enzymatic mechanisms as well as substrate interacting mode than other related MBLs. 19 The side chain of Trp64 that interacts with the hydrophobic side chain of the inhibitor or substrate19,20 is considered to play a role in the movement of this loop. The position Phe70 in NDM-1 has a similar hydrophobic side chain with a suggested similar function. This position was not found in VIM-2 and BlaB (B1 subclass) and CphA (B2 subclass) but showed more similarity to IMP-1. Further, analysis of the primary sequence revealed Tyr2297 to be a unique feature of NDM-1 when compared with universal conserved tryptophan. Tyr229 may not directly participate in processing the enzymatic reaction, as it is obstructed by the floor region. The side chain of this residue forms a hydrogen bond with Gly188 and Leu209; these residues are located at the floor and door region, respectively, indicating their role in active site conformational stabilization. Tyr229 is not conserved with other reported class B1 MBLs but it shows high similarity with class B3 MBL, FEZ-1. 7 Another study suggests the presence of Tyr229 at the periphery of the NDM-1 active site and supports its direct participation in an enzymatic reaction, as confirmed through the Y229W mutational study. 21 Understanding Tyr229 conservation and its functional role is, thus, a matter of further investigation. 16
NDM-1 possesses a unique HAHQD motif when compared with universally conserved HFHDD motif in other MBLs. 7 Substitution of A121F, and Q123D mutations decrease the imipenem hydrolysis activity of NDM-1 by 96.4% and 98.4%, whereas the double mutation A121F/Q123D retained only 0.7% of the wild type activity. As compared with other MBLs, the deep cavity of NDM-1 along with the extraordinary hydrophilic region formed by Trp93, Gln123, Asp124, and His250 provide a larger volume for the enzyme; help them to acquire high binding affinity to cephalosporins, particularly to cefotaxime, cefuroxime, penicillins, and cephalothin. 7
The NDM-1 active site is surrounded by two important loops: A short Loop 3 (residues 67–73) is present in most MBLs B1, and a long Loop 10 (residues 210–230) provides the active site with great flexibility along with facilitating the substrate entrance. 22 In Loop 10, residue L209 forms the hydrogen bond; while interacting with the conserved residue Y229, 23 it creates hydrophobic as well as hydrophilic networks along with the neighboring residues and stabilizes the Loop 10. Moreover, the L209 is C208 after residue, which is a GGC (G206-G207-C208) stretch that acts as a Zn II binding ligand. Due to the presence of two glycine molecules, it allows flexible conformation of C208 that helps the binding of various substrates to the Zn II ion.22,24 Roles of noncatalytic residue Leucine 209 in the NDM-1 active site are explored by changing it into phenylalanine; this substitution was chosen by NDM-1 alignment with common B1 MBLs. Leucine at 209 positions was found to be present in BC II and GIM-1 but does not fall in the conserved region. Using an overlap extension method, 23 laboratory variant L209F of NDM-1 was generated that displayed reduced catalytic efficiency toward penicillin, carbapenems, and cephalosporins. 24 Thus, spatial conformation, along with such unique potential features, is considered a crucial part in deciding the NDM-1 wide spectrum substrate selectivity.
β-Lactamase Mechanism of Action
An important and last step in the synthesis of the bacterial cell wall is peptidoglycan cross-linking, catalyzed by bacterial transpeptidase enzyme (also known as penicillin-binding proteins)25–29
via an active-site serine residue. β-lactamase enzyme acts as a transpeptidase for the synthesis of peptidoglycan. The active-site serine residue of beta-lactamase or transpeptidase enzyme attacks the carbonyl carbon of
Ring-opening mechanism
Sulbactam, tazobactam, and clavulanic acid are some mechanism-based inhibitors that are used in clinics with known β-lactam antibiotics combinational therapies against SBLs. 8 Currently, such efficient mechanism-based inhibitors, either alone or in combination with β-lactam antibiotics against MBLs, are unavailable in clinical settings. 34 The recent addition of NDM to this plasmid-encoded metallo-beta-lactamase along with rapid dissemination hydrolyzes all important clinically available β-lactam antibiotics, including last-generation carbapenems such as imipenem and meropenem.7,35,36 Amide bond cleavage and intermediate protonation are believed to be responsible for the hydrolysis of such β-lactam antibiotics by MBLs. 37 The hydrolysis mechanism begins with the formation of the Michaelis complex (ES) between enzyme and substrate and initiates the first step of amide bond cleavage. In this process, the C-N bond is cleaved by the hydroxide molecule that resides between two zinc ions of the NDM-1 active site.35,38,39 Here, the carbonyl carbon of β-lactam ring is attacked by the nucleophilic action of the water molecule, leading to the opening of the ring along with the generation of an anionic intermediate (EI). Such anionic intermediate decay is a rate-limiting step for the turnover of the antibiotics. They form carboxylation binding with Zn I and amide nitrogen. The carboxylated β-lactam-fused ring interacts further with another zinc ion (Zn II). The amide bond cleavage is followed by a protonation step with a tentatively formed EP complex before it gets released from the enzyme pocket (Fig. 3).35,39,40 The reaction catalyzed by NDM-1 with imipenem, nitrocefin, chromacef, and cefazolin is shown in Fig. 4.

Ring-opening mechanism of β-lactam antibiotic. ES, EI, EP, and P represent enzyme substrate, enzyme intermediate, enzyme product, and product release stages. Source: Feng et al., 2017. 51
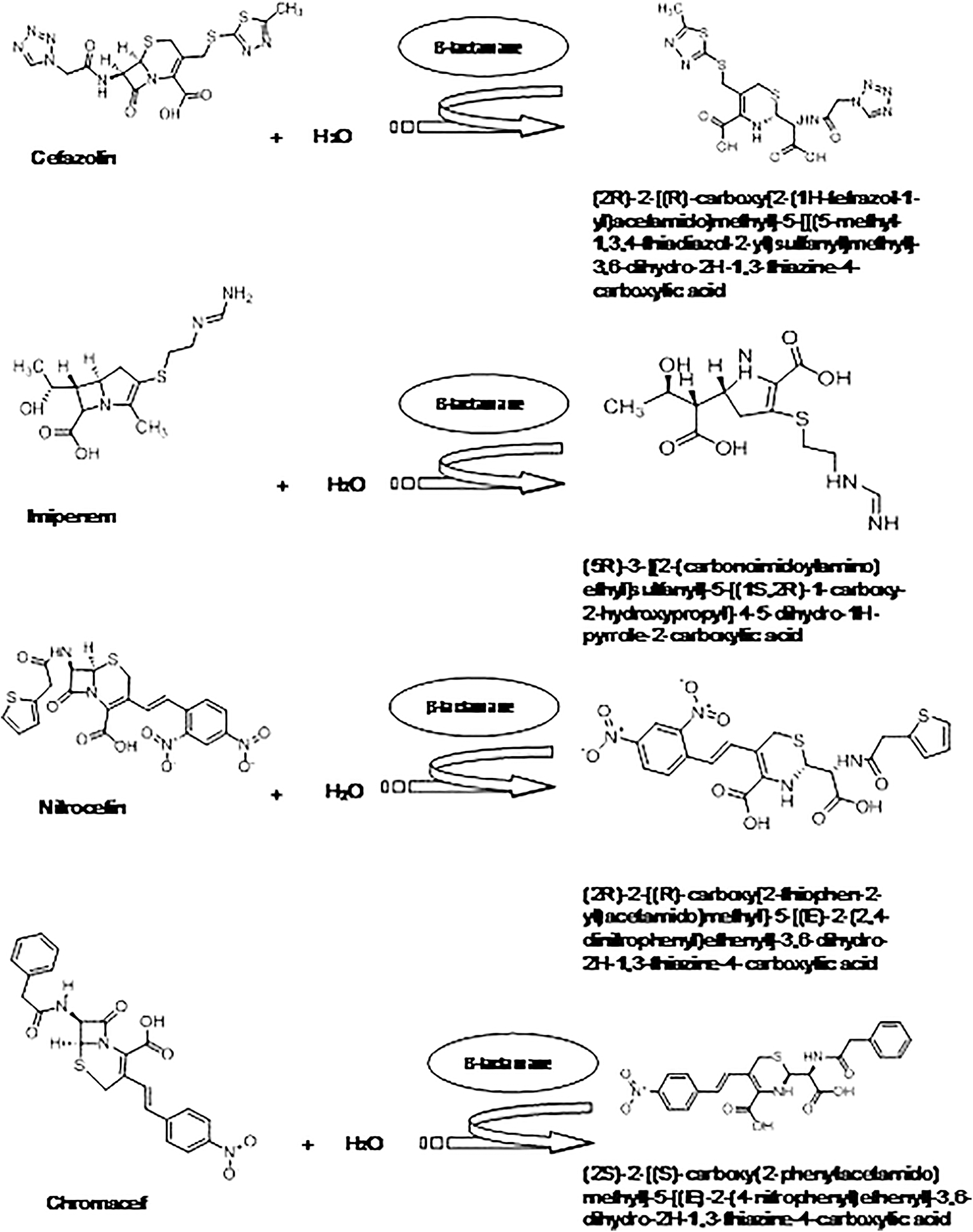
Schematic representation of reaction catalyzed by β-lactamase (E.C. 3.5.2.6). The information is generated by using an enzyme database BRENDA (The comprehensive Enzyme information system).
Two interesting questions after understanding the overall reaction catalyzed by β-lactamase remain unclear. These are: the general mechanism of all hydrolyzable bicyclic β-lactams, and the source for protonation. The source of protonation may be contributed by the bulky solvent, newly formed carboxylic acid or from metal-bound water.41–44 The role of the hydroxide molecule as a proton donor35,38,39 or its bridging role between two zinc ions independent of their distance is well reported in the NDM-1 structure that is complexed with penicillins and cephalosporins.34,35,45 However, such a role is not available in NDM-1 complexed with meropenem (PDB 4EYL and 4RBS). The intermediate formed during penicillin hydrolysis contains a negative charge on the nitrogen of lactam35,39 but in case of cephalosporins or carbapenems, it is delocalized over the conjugated π-system. This encompasses a double bond to lactam-fused dihydrothiazine and even the pyrroline ring and results in the double-bond rearrangement. Such carbon ionic intermediates are reported in various spectroscopic studies where NDM-1 catalyzes the hydrolysis of chromacef, imipenem, nitrocefin, and other B1 MBLs.46–48 The spectroscopic and crystal structure supports pyrroline tautomerization from Δ2 to Δ1 (double bond shift from 2–3 to 3–4) for SBLs-catalyzed carbapenem hydrolysis.49–51 However, there are no such supportive crystallographic studies for MBLs-catalyzed carbapenem hydrolysis. There are reports of two diastereomeric products in BcII-catalyzed imipenem hydrolysis. 47
A recent investigation of X-ray crystallography and nuclear magnetic resonance spectrometry of NDM-1 complexed with meropenem and imipenem supported a different mechanism of carbapenem hydrolysis, particularly in steps involved in the intermediate decay process. Trapping the enzyme intermediate and enzyme product derivatives (EI1, EI2, and EP) in crystal structure revealed the absence of bridging between hydroxide molecules. 51 In addition, double-bond tautomerization and β-diastereomeric product are the distinct features of NDM-1-catalyzed carbapenem hydrolysis. 51 The X-ray crystal structure of NDM-1 in native form or apo form complexed with various substrates or inhibitors is provided in Supplementary Table S1.
Drug Development Based on NDM-1 β-Lactamase Inhibitors
Development of potent inhibitors of NDM-1 is impaired by factors such as shallow active site, lack of inhibitor selectivity,6,52 and lack of novel scaffolds that can selectively target the active site of NDM-1.8,53,54 β-lactam antibiotics or β-lactamase inhibitors in combinations with other NDM-1 inhibitors are available in the literature. These combinations have been successful in combating resistant bacteria-harboring blaNDM-1. 8 Small molecules such as cyclic boronate, 55 thiol-containing carboxylic acids,44,56 some covalent inhibitors such as cefaclor, 57 chromone-3-carboxaldehyde, 58 and natural products 54 are reported as having activity against NDM-1.
A potent broad spectrum inhibitor, VNRX-5133 (taniborbactam), is a new cyclic boronate BLI in clinical development that restored the activity of cefepime against clinical isolates of Enterobacterales and P. aeruginosa with minimum inhibitory concentration (MIC) 1 and 4 μg/mL, respectively. It inhibits both SBLs and MBLs with distinct mechanisms and is the first BLI reported to have direct inhibitory activity against Ambler class A, B, C, and D enzymes. 59
Tables 1 and 2 enlist some lead compounds in the discovery and preclinical phase along with their particular pharmacological action. Information on compounds is sourced from the Clarivate Analytics Integrity database. These compounds were shortlisted based on their pharmacological activity against NDM-1 as having an MIC or inhibitory concentration of 50% (IC50) ≤10 μM.
List of Effective Compounds Reported in Literature (Clarivate Analytics Integrity Database) in Biological Phase
IC50, inhibitory concentration of 50%; MIC, minimum inhibitory concentration; NDM-1, New Delhi metallo-beta-lactamase 1.
List of Effective Compounds Reported in Literature (Clarivate Analytics Integrity Database) in Preclinical Phase
AMA, aspergillomarasmine A; IC50, inhibitory concentration of 50%; MIC, minimum inhibitory concentration; NDM-1, New Delhi metallo-beta-lactamase 1.
Compounds in the discovery phase
Compounds developed based on generating libraries (e.g., 2,6-dipicolinic acid [DPA]) or synthesizing derivatives based on core structure moiety provided efficient inhibitors against MBLs. Utilizing fragment-based drug discovery, a new class of inhibitors is identified against NDM-1, VIM-2, and IMP-1. Libraries synthesized based on DPA were analyzed for structure-activity relationship (SAR); co-administration of a selected inhibitor (IC50 = 80 nM) along with imipenem against clinical isolates of K. pneumoniae and E. coli harboring NDM-1 reduced MIC to susceptible levels. Similar studies were derivatives of cysteine or homocysteine and were shown to be eight times more potent in inhibiting NDM-1 as compared with captopril with IC50 1 μM.60,61
The well-known hypertension drug captopril contains thiol and carboxylate groups that chelate the metal zinc ion in the active site of NDM-1. Both D and L captopril stereoisomer have been reported to inhibit NDM-1 with IC50 values of ∼8 and ∼200 mM, respectively.16,44 An example of thiol-based MBL inhibitors, thiomandelic acid and 2-mercapto-3-phenyl propionic acid was found to resensitize the imipenemase-producing K. pneumoniae strain, when used in synergy with meropenem. 62 Five novel non-β-lactam chemical compounds (AW01220, BTB02323, RF01991, AW01120, and HAN00044) virtually screened from Maybridge database having high GOLD fitness score and binding energies compared with mercaptocarboxylate, metal enzyme inhibitors are reported.36,63 In the molecular dynamic simulation study, microbiological and kinetics parameters confirmed these compounds as highly suitable to be used as potential future drug candidates, with all compounds showing IC50 in the nano molar range ≤2.2 nm.36,63
The compounds in the discovery phase are categorized into various groups based on their structural core moiety. Such grouping provided us with a total of 30 compounds grouped into 6 classes, including (2-oxoalkyl)thio, Isatin, Imidazo-thiazolylhydrazinocarboxamide, Biphenyl carboxylic acid, Mercaptophosphonate, and dihydro-1H-imidazole moiety containing compounds and a few compounds with miscellaneous structure.
(2-Oxoalkyl)thio-based NDM inhibitors
The (2-Oxoalkyl)thio-based NDM inhibitors represent 12 molecules in this series. Among them, three molecules (Integrity Entry No. 887340–887342) 64 have an MIC of 4 μg/mL. The essential requirement of molecules such as the beta-lactamase NDM-1 inhibitor is to interact with binding sites, that is, Zn I and II and lysine residue 221. The docking studies for entry numbers (887340–887342) reveals that the triazole group binds to the Zn I and II and the amide portion interacts with the Lys 211, which blocks the active site and prevents the cleavage of the lactam ring. In another study, the oxoalkyl(thio)-based NDM inhibitor was synthesized, and the results (Integrity Entry Nos. 920674, 920675 and 920678) were highly potent with an IC50 value of <1 μM. 65 Here also, the binding interaction was studied (983335); the substitution affects the binding of molecules with the main domain of the active site as compared with the previous study. In this study, the nitro group of aromatic portion bridging with the Zn II site and amide portion interacts with the lysine 211.
Isatin-based NDM inhibitors
(Z)-2-(1-methyl-2-oxoindolin-3-ylidene)hydrazinecarbothioamide: a new class of isatin, 69 that is, 1H-indole-2,3-dione indole derivative, an endogenous natural product (Integrity Entry Nos. 796806 and 796808) was reported as NDM-1 inhibitors, with IC50 3.30 μM and 6.29 ± 0.940 μM, respectively. Substitution of I or bromo group on isatin moiety increased the pharmacological activity compared with already reported methisazone (structure not shown). One such compound from this substitution (Integrity Entry No. 400031) showed an IC50 value of 298 μM against E. coli beta-lactamase NDM-1. Docking studies revealed that the thiol group present in the molecule forms several coordination bonds with zinc ions, suggesting their essential roles in the inhibition process. In addition to this, many residues of β-lactamase interact via hydrophobic interactions with different amino acid residues (Thr34, Val73, Trp93, Gly219, Asn220, and Gly222). In addition to that, two hydrogen bonding interactions with Gly69 and His189 are also reported.
Imidazo-thiazolylhydrazinocarboxamide moiety-based inhibitors
The Imidazo-thiazolylhydrazinocarboxamide moiety-based NDM inhibitors exhibit a potent activity of IC50 0.600 and 0.290 μM. Integrity Entry numbers 901119 and 901122 represent the Imidazothiazolylhydrazinocarboxamide moiety-based NDM inhibitors, where replacement of “O” with “S” improved the activity 70 (Table 1). The docking studies for the compounds in this class reveal that the sulfur residue of molecules binds with the Zn I binding site, and the aromatic portions such as the phenyl ring displace the other active site Zn II accompanied by the reorientation of the groups that originally chelated with the Zn.
Biphenylcarboxylic acid moiety-based NDM inhibitors
This class of inhibitors representing the biphenylcarboxylic acid moiety-based NDM inhibitors (Integrity Entry Nos. 994343 and 994345) exhibited IC50 values of 0.310 and 0.570 μM, respectively. 72 The docking studies of these molecules revealed that the thiol portion interacts with the Zn I and II binding site, and the acid portion interacts with the lysine 221. Thiols have the potential to coordinate with the zinc ions in the MBL active site. This thiophilic nature of zinc prevents β-lactam hydrolysis.
Mercaptophosphonate-based NDM inhibitors
(R)-methyl 2-(((benzyloxy)carbonyl)amino)-3-mercaptopropanoate: This category of NDM inhibitor-containing mercaptophosphonate forms the basic core moiety. Integrity Entry numbers 965998, 965999, and 966000 belonging to this class displayed potent IC50 values of 1.81, 6.60, and 2.50 μM, respectively. 71 This class also has a thiols group that coordinates with the zinc ions in a way similar to biphenylcarboxylic acid moiety-based NDM inhibitors. Here, Asn 233 residue of NDM-1 showed an additional interaction with the P-O group.
Dihydro-1H-imidazole moiety-based NDM inhibitors
The dihydroimidz-1H-imidazole moiety-based NDM inhibitors are represented by eight molecules exhibiting an IC50 ranging from 0.4 to 1.7 μM (Table 1 with S. Nos. 22–29). The SAR from these structures revealed that the bulkier group attached to the basic core on all sites and the presence of the halo group that is, the chloro or bromo group showed good activity. There was no effect in the activity by replacement of the chloro group with the methoxy or aryl group. 73 This is the new class of inhibitors whose binding details are yet to be explained.
Compounds in Preclinical Phase of Development
A fungal natural product Aspergillomarasmine A (AMA) has gone into an advanced phase of preclinical development. AMA (Integrity Entry No. 855642) inhibited NDM-1 (IC50 = 4.0 μM) 54 ; AMA in combination with meropenem was found to be effective in both in vitro and in vivo studies. 54 It fully restored the bactericidal property of carbapenems against Acinetobacter, Enterobacteriaceae, and Pseudomonas spp. possessing either NDM or VIM-type alleles. An in vivo study demonstrated AMA (Integrity Entry No. 855642) as a nontoxic adjuvant candidate that restored meropenem activity when tested in mice infected with NDM-1-expressing K. pneumoniae. Its inactivation mechanism remains consistent with known in vitro chelators of B1 MBLs subclass that involve the removal of Zn2+ ions from the active site. 54 A compound library based on 1,2-benzisoselenazol-3(2H)-one derivative (Integrity Entry No. 1014299) provided a novel nontoxic scaffold as an adjuvant efficiently enhancing the meropenem antibacterial activity against NDM-1-producing carbapenems-resistant Enterobacteriaceae clinical isolates. When used in combination, it effectively restored the carbapenem activity against the in vivo Galleria mellonella larvae infection model. The mode of inhibition of this compound on the purified enzyme depicted its covalent binding and displaced one zinc ion from an active site. 78
Molecular dynamic simulation or mutational analysis study revealed the interaction of antibiotics with the active site of NDM-1. Antitumor medicine embelin, when tested against beta-lactamase NDM-1 carbapenemase, exhibited a value of IC50 2.1 ± 0.2 μM. Its molecular dynamic simulation revealed an interaction of embelin's hydroxyl group with the Zn2+ metal ion molecule buried within the active site. This antitumor drug molecule appeared as a promising carbapenem adjuvant in restoring the meropenem activity when tested against a panel of NDM-1 harboring A. baumannii, E. coli, and K. pneumonia. 79 Mutational analysis and molecular modeling studies demonstrated the binding of magnolol (a natural compound isolated from magnolia bark) at 110–200 residues of the catalytic pocket, thereby blocking the binding of the substrate and leading to its inactivation. At an IC50 value of 6.47 μg/mL, it restored the meropenem activity against E. coli ZC-YN3. 80 Colistin re-emerged as an effective antibiotic against multidrug NDM-1 harboring bacteria but due to its nephrotoxicity effect its use was restricted and dosage optimization became a need. A recent study where succinic acid, or oxalic acid was tested in combination with colistin not only potentially reduced the MIC of colistin from 8 to 4 μg/mL but also reduced its dosage to 0.5 μg/mL and made it safer to be used for clinical application. 81
Another novel inhibitor that provides an opportunity to develop effective combination therapy is ANT431 (Integrity Entry No. 871911). In vivo proof of concept demonstrated the nullifying effect of the compound on E. coli clinical isolates expressing MBL in the mouse infection model. 77 In addition to compounds mentioned in Table 2, Drug Dipi-Van or Dipicolyl-vancomycin conjugate (Integrity Entry No. 931528) (Fig. 5) is in preclinical phase targeting NDM-1. It has antibacterial activity against K. pneumoniae, E. coli, Staphylococcus aureus, Enterococcus faecium, Enterococcus facecalis etc. with an MIC range of 0.3–12 mg/L. 82 This conjugate of vancomycin also restored the activity of meropenem against NDM-1-producing clinical isolates of Gram-negative pathogens such as K. pneumoniae and E. coli. This rationally developed inhibitor can penetrate the outer membrane of Gram-negative pathogens and inactivates NDM-1 enzyme by depleting the metal ion (Zn2+) from its active site. Further, the restoring activity of the inhibitor is efficiently mentioned against NDM-1-producing K. pneumoniae in the murine sepsis infection model. 83
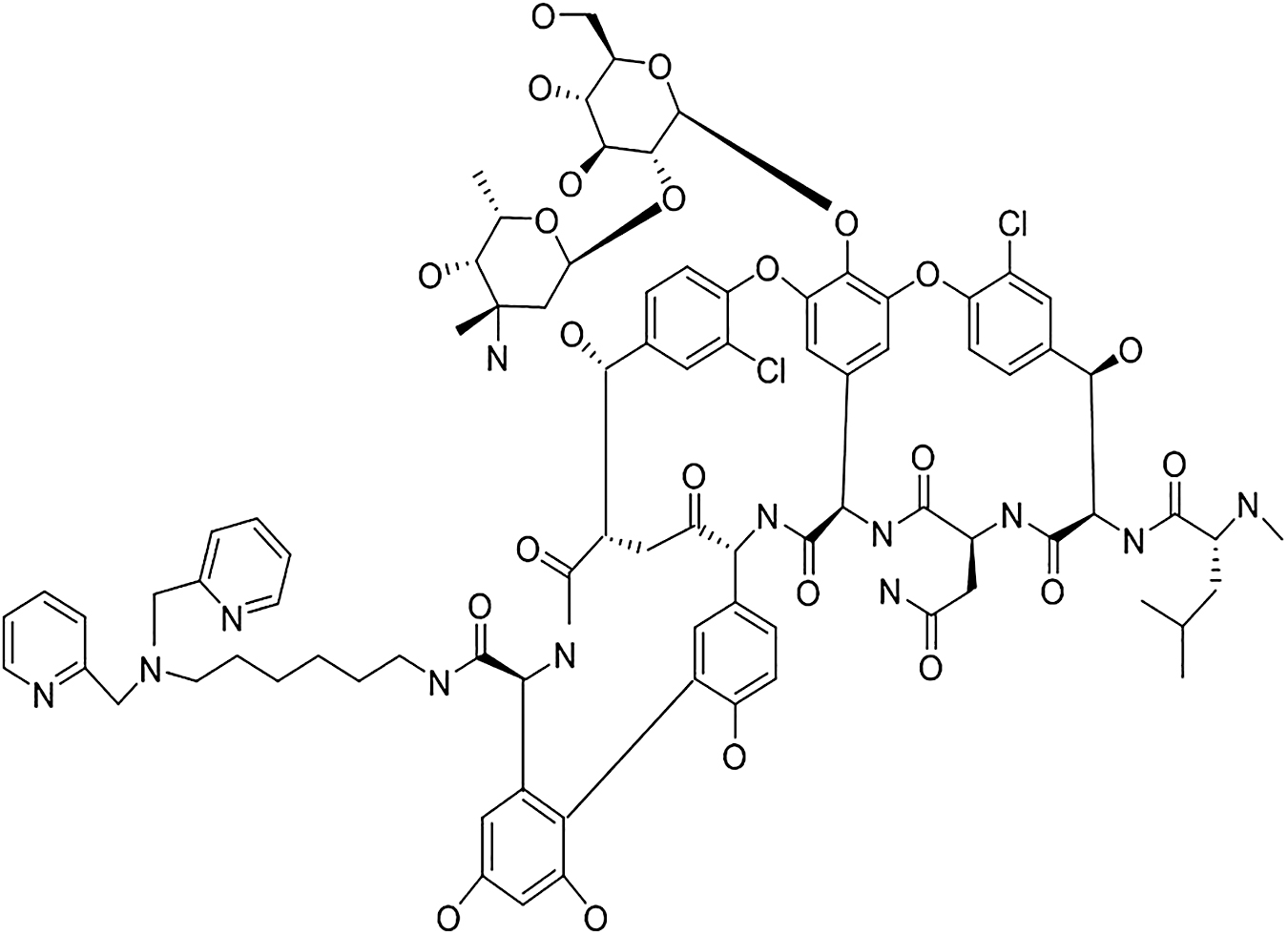
Shows structure of drug Dipolyl-vancomycin conjugates (Integrity Entry No. 931528).
Use of metal chelators (e.g., trispicolylamine [TPA]-based zinc chelators) is also reported to rescue the β-lactam antibiotics from cleavage by NDM-1. Hydrolysis of the β-lactam ring of antibiotic is brought about by the presence of the Zn2+ coordinated water molecule in the NDM-1 active site. Removing zinc ions from the active site by using zinc chelators as a potent adjuvant with higher degree chelation selectivity that does not affect host metalloenzymes ensured MBLs inhibition and restored the antibiotic property.84–87 Selective zinc TPA (Integrity Entry No. 1017703) (Fig. 6) based synthesis of chemical compounds with an addition of linker and modulator restored the bactericidal activity of meropenem when used in combination against P. aeruginosa and K. pneumoniae expressing carbapenemases NDM-1 and VIM-2. Further, in vivo toxicity in the mice model showed no acute toxic effect even at a dose of 128 mg/kg. 88

Shows structure of the compound in preclinical phase (Integrity Entry No. 1017703).
The generalized mechanism of the action of compounds/inhibitors mentioned here shows covalent binding and displacement or depletion of the zinc ion from its active site in a way that in vitro chelators do. Further, the interaction of the inhibitors with binding sites, that is, Zn I and Zn II and lysine residue 221 blocks the active site, thus, preventing lactam ring cleavage. Table 3 summarizes NDM-1 inhibitors based on the mechanism of action of NDM-1 inhibitors.
Mechanism of Action of NDM-1 Inhibitors
AMA, aspergillomarasmine A; NDM-1, New Delhi metallo-beta-lactamase 1.
In addition to the mentioned drugs, a synthesized peptide is also reported to inhibit NDM-1. One such example is the use of a peptide (RXR)4 comprising (Arg-6-aminohexanoic acid-Arg) 4 peptide conjugated to 11-mer phosphorodiamidate morpholino oligomers (PPMOs). It is reported to be in the preclinical phase by Trijin Medical University (Integrity Entry No. 849590/code name M12-PMO). Another antimicrobial peptide, thantin, is reported to reverse carbapenem resistance in NDM-1-producing bacteria both in vitro and in vivo. With the dual mechanism of action, it combats the infections caused by NDM-1-producing pathogens by displacing the zinc ions from the active site and the competitive displacement of divalent cations that induce the release of lipopolysaccharides, which leads to disrupting the outer membrane of bacteria. 90 Researchers also describe M12-PMO conjugated with morpholino oligomers as a practical way that potentially treated Duchenne muscular dystrophy and an alternative to currently available antisense oligonucleotides. 91
Clinical studies
There is a report of a successful treatment of kidney transplant recipients suffering from urinary tract infection caused by multidrug-resistant K. pneumoniae-producing NDM-1 strains. These patients were given Co-trimoxazole 2 × 960 mg/day for 3 weeks followed by 480 mg/day, and 3 doses of fosfomycin prophylaxis. 92
Conclusion
Targeting or interfering with cell wall synthesis is an effective and fast way of disturbing bacterial cell viability that ultimately leads to cell death. NDM-1, a recently emerged MBL class, is disseminating within the microbial population. This MBL class is responsible for hydrolyzing all important clinically available β-lactam antibiotics, including last-generation carbapenems (imipenem and meropenem). The inhibitors described here are primarily zinc ion binding inhibitors. These inhibitors include zinc ion binding inhibitors that form ternary [MBL-ZN(II)-inhibitors] complexes and zinc stripping inhibitors that result in a zinc-inhibitor complex and an MBL with reduced amounts of Zn(II) bound. Utilizing such metal-binding inhibitors to overcome MBL-mediated resistance looks promising, as the presence of the metal center in the active site of the enzyme is its defining feature as well as constitutes the most conserved region of such a diverse family of enzymes. Some of these inhibitors have entered the preclinical phase of development. It was difficult to draw a general pharmacophore for the mentioned compounds but still the minimum structural requirements for a molecule to inactivate NDM-1 are: (1) the presence of a hetero atom (i.e., N or S) for interaction with zinc ions; (2) the presence of a hydrogen bond acceptor to interact with one of the key amino acid residues (Lys211) present in the active site.
Working on a peptide-based linker that specifically targets cell wall synthesis also seems to look a promising way to target bacterial killing. Further, the combination therapy with a novel compound-developed chemistry leading to optimization has therapeutic potential that can address the issue of challenges presented by MBLs-producing resistant strains. Improving the biological efficacy of inhibitors through selective chelators with a modular and a compatible linker serves as a potent adjuvant that enhances and even restores the bactericidal activity of β-lactam antibiotics.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.