
Abstract
Many members of nontuberculous mycobacteria (NTM) are opportunistic pathogens causing several infections in animals. The incidence of NTM infections and emergence of drug-resistant NTM strains are rising worldwide, emphasizing the need to develop novel anti-NTM drugs. The present study is aimed to identify broad-spectrum drug targets in NTM using a comparative genomics approach. The study identified 537 core proteins in NTM of which 45 were pathogen specific and essential for the survival of pathogens. Furthermore, druggability analysis indicated that 15 were druggable among those 45 proteins. These 15 proteins, which were core proteins, pathogen-specific, essential, and druggable, were considered as potential broad-spectrum candidates. Based on their locations in cytoplasm and membrane, targets were classified as drug and vaccine targets. The identified 15 targets were different enzymes, carrier proteins, transcriptional regulator, two-component system protein, ribosomal, and binding proteins. The identified targets could further be utilized by researchers to design inhibitors for the discovery of antimicrobial agents.
Introduction
Over the past two decades, emerging and re-emerging infectious diseases have started posing a severe threat to the otherwise won-over battle of the humans against the bacterial pathogens. 1 This threat can be partially attributed to the evolution of the pathogens, due to the acquired drug resistance and the inadvertent usage of the antibiotics, as exemplified by the disease tuberculosis and malaria. However, other factors, like inadequate vaccination and poor immunity of the patients, have also led to the re-emergence of diseases like diphtheria and whooping cough (pertussis). In fact, immunocompromised cases have contributed heavily to the development of a number of diseases caused by the opportunistic pathogens (OPs). 1 Ideally, these OPs are potentially dangerous enough to cause infectious diseases, sparing only the healthy individuals with strong and sound immune systems. Nontuberculous mycobacteria (NTM), incidentally, happen to fall into this category of OPs and are known to be continuously rising, thereby signifying a growing public health problem.
To date, more than 190 distinct Mycobacterium species have been unearthed. 2 These comprise both the pathogenic ones, like the well-known and clinically important Mycobacterium tuberculosis and Mycobacterium leprae, as well as the nonpathogenic ones like Mycobacterium smegmatis. 2 Among these, around 150 belong to the NTM group 3 that do not cause tuberculosis as well as leprosy. 4 NTM species are ubiquitously found in the environment, mainly in soil and water 2 , along with ∼10% of cystic fibrosis patients. 5 In fact, some NTM species frequently cause severe pulmonary infections (65–90%) in humans3,4 in addition to infecting skin, soft tissue, lymph nodes, bone, blood, and other usually sterile locations in the body, primarily in immunocompromised patients, as reported by the Centers for Disease Control and Prevention (CDC). 6 NTM infection rate alarmingly increased from 1.3% to 32.7% in patients diagnosed with cystic fibrosis, when left untreated.7,8 Most common drug regimens to treat these emerging infectious diseases caused by NTM include macrolides (i.e., clarithromycin and azithromycin) and quinolones (i.e., ciprofloxacin and ofloxacin). 9 However, such treatment options have been restricted by the high incidence of drug resistance among NTM species.10–12 This intrinsic drug resistance is generally conferred by the drug efflux systems, cell-wall permeability and low-affinity drug targets besides the additionally acquired resistance developed due to chromosomal mutations. 13 Moreover, the NTM antibacterial drug development has witnessed a lethargic growth in the past few decades. 14 Thus, the current scenario emphasizes the need for improved therapeutic interventions to prevent and control NTM infections worldwide.
With an aim to develop novel classes of antimicrobial agents, exploring new and additional drug target proteins can be of great help.15,16 In fact, computational approaches, encompassing but not limited to network analytical approaches, for the identification of therapeutic candidates,16–19 are much cheaper, as well as faster, compared with experimental methods. Among other computational methods, comparative and subtractive genomic approaches have been widely used to identify drug targets in numerous pathogens.20–22 Both the approaches utilize the criteria of essentiality and selectivity/specificity for predicting therapeutic candidates. Comparative genome analyses have been used for drug discovery research in Mycobacterium species, 23 including M. tuberculosis, 24 M. leprae, 25 Mycobacterium abscessus, 26 and Mycobacterium ulcerans. 27
Despite several reports on different species of Mycobacterium on the utilization of the genomics and bioinformatics approaches for the identification of new drug targets, reports on the identification of broad-spectrum target candidates for NTM are scanty. For instance, comparative analyses of the genomes along with the proteomes and phylogenies of 21 tuberculosis and nontuberculosis strains performed by Zakham et al. revealed considerable conservation in mycobacterium gene families. 28 Again, comparative analyses of various published genomes of M. abscessus complex (MAC), carried out by Sassi and Drancourt, 29 reported the presence of three genomospecies (M. abscessus, Mycobacterium bolletii, and Mycobacterium massiliense), to facilitate the understanding of their pathogenesis factors, and unravel novel specific targets for drug design and diagnosis tools. Furthermore, the comparison of 10 mycobacterial genomes, including the pathogenic and nonpathogenic species, reported that the predicted drug targets for M. tuberculosis have counterparts (orthologs) in other mycobacterial species and thus these targets can be used for treating other mycobacterial infections. 30
In the present study, we exploit the comparative genomics approach to gradually shortlist a set of broad-spectrum targets in a wide range of NTM. We have identified 15 broad-spectrum targets in 24 NTM based on the criteria of essentiality, selectivity, and druggability.
Materials and Methods
In the present study, a total of 24 NTM species were considered for the identification of broad-spectrum targets. NTM species were selected on the basis of (1) the availability of their complete genome sequences (until June 2017) and (2) clinical reports of causing opportunistic infections in the human population (cited in Table 1). The overall methodology followed in this study is depicted in Fig. 1.
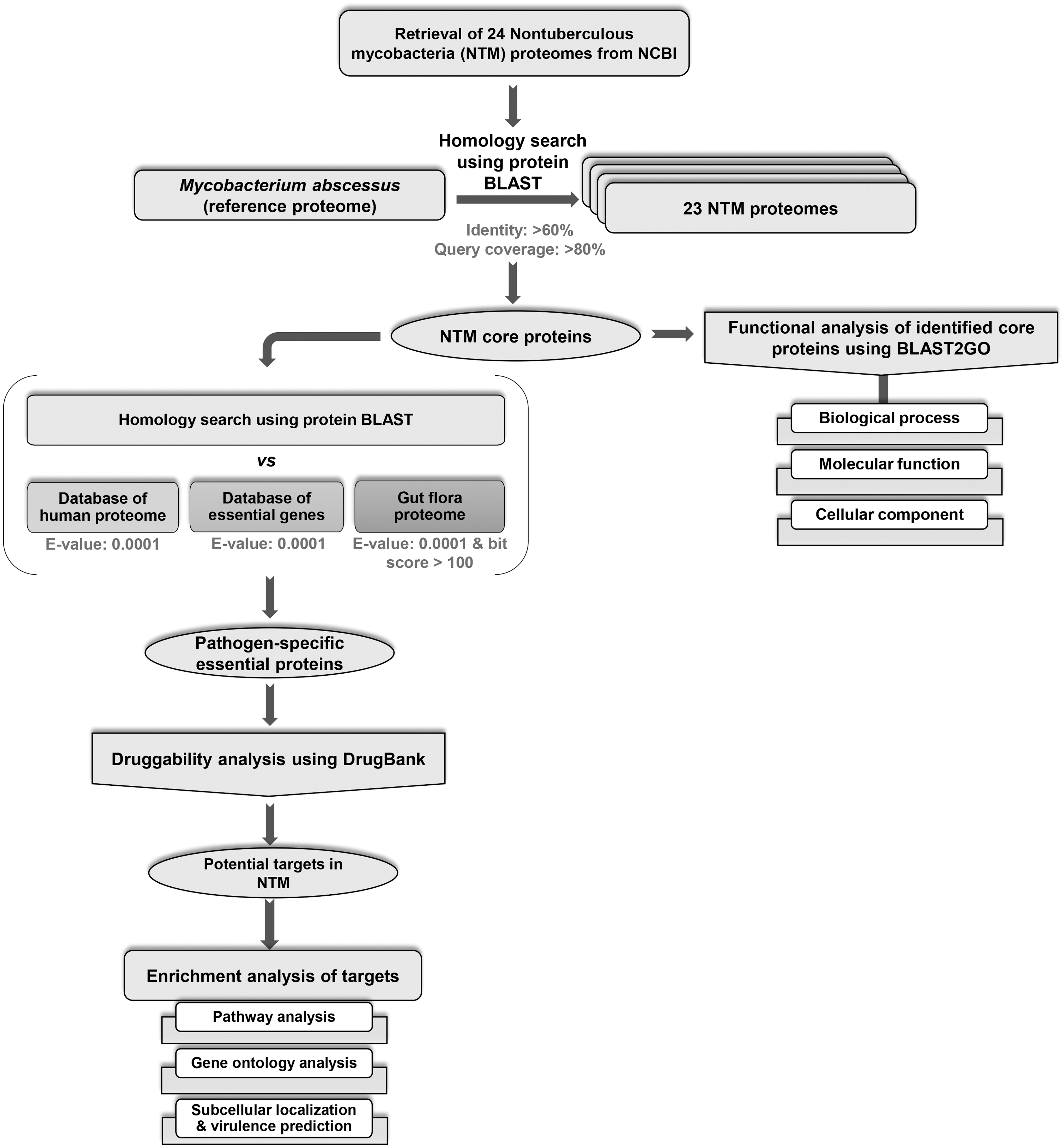
The workflow adopted in the present study.
Information of Pathogenic Nontuberculous Mycobacteria Considered in the Study
NTM, nontuberculous mycobacteria.
Identification of core proteins
The proteome sequences of 24 NTM species were retrieved from the NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/bacteria). A pan-proteome analysis was performed using standalone BLAST 31 to compare the proteomes of NTM across the different species. This analysis would help to understand the functional conservation and diversity of proteins and thereby identify the core/accessory proteins present in all NTM under consideration. The core proteins were filtered with >60% sequence identity and >80% query coverage. 32 For such comparison, M. abscessus was considered as the reference proteome as the former is a part of the MAC, which comprises various fast-growing mycobacterium species causing a wide range of infection in clinical environment, including soft skin tissue infection of wounds and pulmonary infection in cystic fibrosis patients.
Functional analysis of core proteins
The identified core NTM proteins were analyzed using BLAST2GO, a bioinformatics suite for functional analysis and annotation of gene/protein sequences. BLAST2GO annotates the query proteins by initially finding the homologous sequences using BLAST and later mapping the gene ontology (GO) terms to the obtained hits, thereby, providing a comprehensive annotation result. 33
Shortlisting pathogen-specific and essential proteins
The core proteins, resulting from the pan-proteome analysis, were subjected to a homology search using protein BLAST (BLASTp) against the nonredundant (nr) database of the human proteome with an e-value threshold of >0.0001. The proteins, which showed no significant hits with human proteins, were considered for further analysis. To assess the essentiality of these proteins, a homology search against the bacterial proteins from the Database of Essential Genes (DEG) was carried out using e-value <0.0001 and a bit score >100 as cutoff value. DEG is a repository of experimentally determined essential genes of different bacteria, archaea, and eukaryotes. 34 Furthermore, shortlisted NTM proteins were compared with gut flora proteomes using BLASTp search, and NTM proteins, satisfying the criteria of e-value >0.0001 and bit score <100, were selected.22,35 Gut flora proteomes from Jadhav et al 22 was used in the present study for the identification of nongut flora proteins.
Druggability analysis
Druggability of a protein describes its ability to bind to small drug-like molecules with high affinity. Here, druggability of the shortlisted pathogen-specific essential proteins was analyzed by carrying out a homology search against the targets from the DrugBank database, a repository of detailed information about drugs and their targets, 36 with default parameters. Proteins with a high percentage of identity to targets present in DrugBank (e-value <0.0001) are likely to be druggable.
Enrichment analysis of shortlisted proteins
Pathway enrichment and GO analysis of shortlisted druggable proteins were performed using bioinformatics resources, namely KEGG, DAVID, and CELLO2GO. KEGG is a comprehensive database of genomes, biological pathways, diseases, and drugs. 37 DAVID is an integrated knowledge database for annotation, visualization, and integrated discovery. 38 CELLO2GO is a bioinformatics tool used for analyzing protein properties along with their subcellular localizations. 39
Subcellular localization and virulence prediction
Further in this study, the subcellular localization and virulence factors among the shortlisted proteins were predicted using PSORTb, MP3, and VirulentPred. PSORTb is a web-based subcellular localization prediction tool for bacteria and archaea with an accuracy of ∼98%. 40 VirulentPred is a support vector machine (SVM)-based online server for bacterial virulence factor prediction, 41 whereas MP3 is an SVM-hidden Markov model-based tool to predict pathogenic proteins in both genomics and metagenomics datasets. 42
Results and Discussion
In the present study, the comparative genomics approach enabled us to prioritize broad-spectrum antimycobacterial targets in NTM. The analysis involved (1) identification and functional analysis of core proteins present in all NTM taken for this study, (2) identification of pathogen-specific and essential proteins indispensable for their survival, (3) druggability analysis, and (4) enrichment analysis of potential target proteins, including subcellular localization and virulent prediction.
Identification and functional annotation of core proteins
A total of 1,19,233 protein sequences from 24 NTM were collected from NCBI (Table 1). Using M. abscessus proteome as a reference in pan-proteome analysis, 537 common proteins were identified from the NTM protein dataset (Supplementary Table S1). These common proteins (orthologous proteins), present across the diverse NTM species, are believed to be evolutionarily conserved and are referred to as the core proteins. To further elucidate the role of these core proteins, functional annotation was carried out using BLAST2GO, a tool that annotates the proteins with respect to their cellular component, molecular function, and biological processes. Functional annotation of the core proteins revealed that these proteins were located mainly in the membrane, followed by cytosol, cell wall, and cytoplasm (Fig. 2). Molecular functional analysis showed that the majority of the core proteins were involved in ATP binding and protein-binding processes, while some others showed a significant role in biological functions like magnesium ion binding and DNA binding. The oxidation/reduction process, translation, pathogenesis, and response to the antibiotics were the predominant biological processes in which the core proteins were involved (Fig. 2). Participation of core proteins in such vital biological processes is an important feature to develop novel anti-NTM drugs.
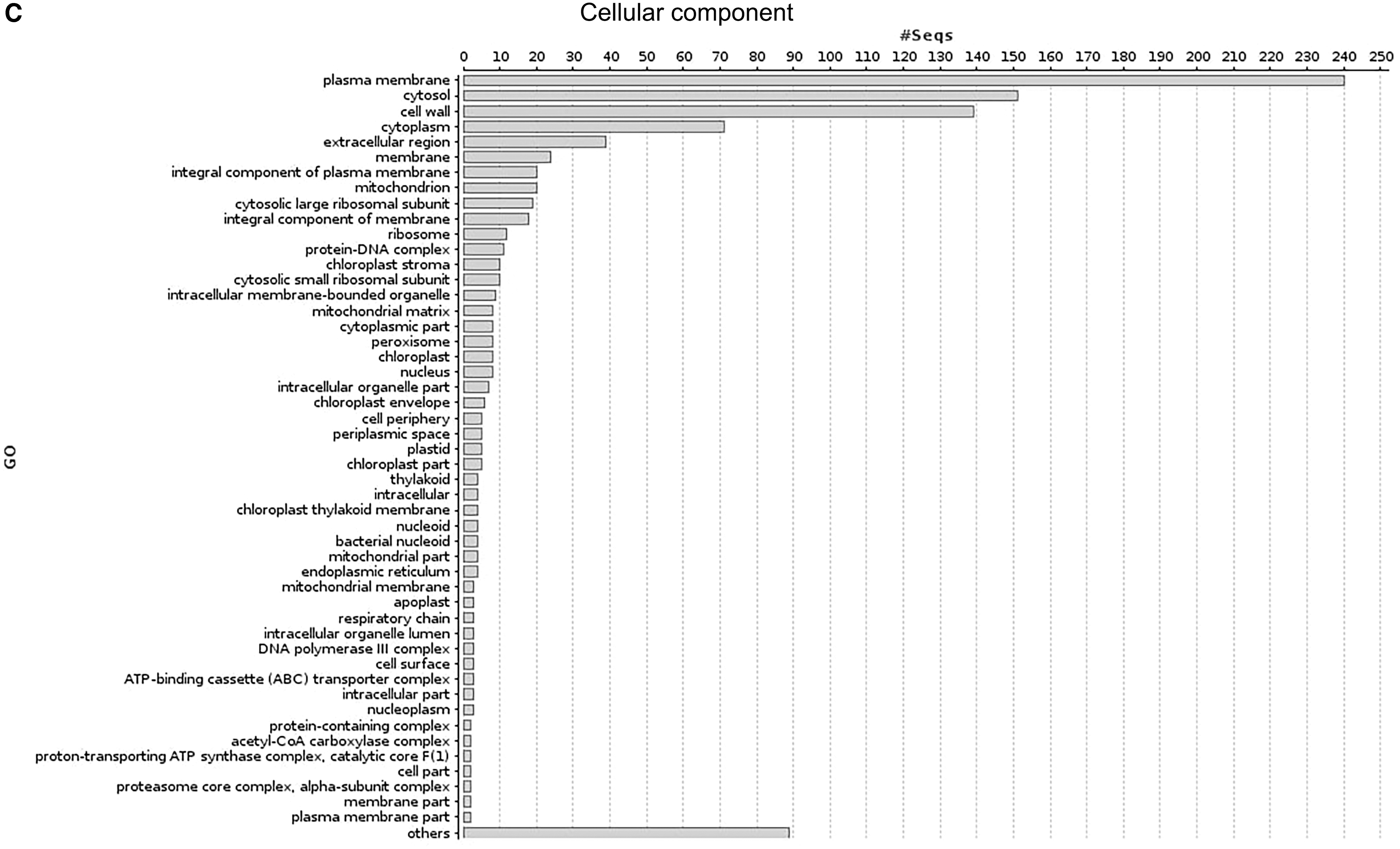
Distribution of enriched gene ontology terms for the identified nontuberculous mycobacteria core proteins showing the top hits.
Identification of essential and pathogen-specific proteins
Ideally, to minimize the undesirable host/drug interactions, a drug target that showed minimum (or no) similarity with host protein was considered for the study. Thus, the NTM core proteins were compared with the human proteome. It was observed that among 537 core proteins, 222 did not show any similarity with the human proteome and thus, the later were considered specific to NTM species. Likewise, the filtering of proteins, which were proposed to be essential for the growth and survival of the organism, could yield better drug targets in terms of their potency. Proteins, which are similar to the known essential proteins, are likely to be essential. A similarity search against DEG was, therefore, carried out to identify the crucial proteins for the growth and survival of NTM. Among the previously shortlisted 222 proteins, 173 showed similarity to those proteins present in DEG and thus, these were considered as essential proteins (Supplementary Table S2).
Additionally, an ideal target should not have similarity with human gut flora proteins to avoid any impediment in the functioning of the beneficial microbiota proteins. Indeed, an enormous number of beneficial microorganisms (i.e., gut flora or microbiota) reside in the human intestine. These gut flora play crucial roles in protecting hosts against pathogenesis by regulating the immune system. 43 Thus, a screening of 173 NTM proteins was carried out by comparing with beneficial gut flora proteomes (with e-value >0.0001 and bit score <100) to select proteins that are nonhomologous to gut flora proteins. This resulted in 45 proteins (Supplementary Table S3) as nonhomologous to gut flora. Finally, these 45 proteins were considered to be essential for the survival of NTMs and nonhomologous to human and gut flora proteins, which were taken for further analysis.
Identification of druggable candidates
Druggability, the possibility of being able to modulate a target with a small-molecule drug, is one of the important parameters for screening therapeutic targets. Proteins that show high similarity with known drug targets can be considered as druggable targets. Of the shortlisted 45 NTM proteins, 15 proteins showed homology with known targets in DrugBank database and were considered as druggable (Table 2), and the remaining 30 proteins as novel drug targets, which are worth further exploration. Some of the identified druggable targets were found to be present in other pathogens, including Streptococcus sanguinis, Streptococcus agalactiae A909, and M. tuberculosis H37Rv.
List of Identified Broad-Spectrum Targets in Nontuberculous Mycobacteria
TetR, Tet repressor.
In the present study, the identified 15 core proteins, which were pathogen-specific, indispensable for growth and survival as well as druggable, were considered as promising broad-spectrum target proteins. DAVID and KEGG analyses revealed that some of these proteins were engaged in vital metabolic pathways, such as lysine biosynthesis, citric acid cycle (TCA), starch, and sucrose metabolism, oxidative phosphorylation, etc. (Table 2). Among these 15 proteins, CELLO2GO predicted 13 to be located in the cytoplasm, 1 in extracellular matrix, and 1 in membrane whereas PSORTb predicted 13 proteins as cytoplasmic and 2 as unknown (Table 2). Ideally, cytoplasmic and membrane proteins could be considered as good drug and vaccine targets, respectively. Also, a virulent protein prediction using VirulentPred revealed that among the 15 candidate proteins, six were virulence factors, whereas MP3 predicted three proteins as virulent in nature (Table 2). Two proteins were commonly identified as virulent factors by both the prediction methods.
Identified broad-spectrum targets in NTM
The series of in silico analyses led to the identification of 15 broad-spectrum targets as drug/vaccine candidates in NTM species (Table 2). The target proteins belong to various functional classes, which are discussed below.
Hydrolase (MAB_1067)
In the bacterial kingdom, hydrolases are a predominant class of biocatalysts, which carry out important degradative reactions by cleaving large molecules into fragments. 44 Furthermore, hydrolases are also involved in several critical functions like cell wall growth, peptidoglycan maturation, elongation turnover, recycling, autolysis, and the separation of daughter cells during cell division in bacteria. 45 In the present study, the proposed target (MAB_1067) is a haloacid dehalogenase-like hydrolase. Several hydrolases have been reported as drug targets in various pathogenic bacteria, including M. tuberculosis, Acinetobacter baumannii, and Helicobacter pylori.46–48
RimJ (MAB_1088)
RimJ is an alanine acetyltransferase, which acetylates the α-amino group of the N-terminal alanine residue of ribosomal protein S5, a component of 30S ribosomal unit. It plays an important role in the maturation of the 30S ribosomal subunit. 49 Ribosomal proteins are important components of the ribosomal subunits involved in protein biosynthesis. Hence, inhibiting these acetyl-transferases would cause major damage to bacteria, making them an excellent potential drug target. Silvério-Machado et al. identified ribosomal-protein-alanine acetyltransferase as a potential target candidate within the members of the Enterobacteriaceae family using singular value decomposition method. 50
Transcriptional regulator, Tet repressor family (MAB_1149)
TetA is a membrane-associated protein that exports antibiotics out of bacterial cells. The expression of TetA is regulated by Tet repressor (TetR) family of proteins. 51 TetR family of regulators (TFRs) have an N-terminal DNA-binding domain and a large C-terminal domain. 52 C-terminal domain binds tetracycline and upon binding of tetracycline with TetR, a conformational change occurs, which blocks the binding of TerR with DNA. This conformational change is responsible for the regulation and induction of tetracycline resistance. TFRs are involved in regulating catabolic pathways and transcriptional control of multidrug efflux pumps. These regulators also respond to specific environmental conditions, such as, osmotic stress and cellular signals, which modulate gene expression levels. 53 In M. tuberculosis, a TetR family of DNA-binding protein, which is involved in antibiotic resistance, has been identified as a possible drug target. 54
Glycogen phosphorylase, Glgp (MAB_1469)
Glycogen phosphorylase (GlgP) is a glucan-degrading enzyme that catalyzes the glycogen breakdown by the reversible cleavage of α-1,4 bond at the nonreducing ends of polyglucans resulting in glucose-1-phosphate. 55 GlgP (EC 2.4.1.1) is the key enzyme of carbohydrate metabolism that helps in the biosynthesis of secondary metabolites. It is reported that the activity of GlgP is regulated by the phosphorylation status of histidine-containing phosphocarrier protein (HPr, a phosphotransferase system). 56 HPr is primarily in the dephospho form, which leads to the activation of glycogen phosphorylase and degradation of glycogen. This degraded glycogen serves as an extra source of energy during the early stages of growth. GlgP along with GlgX (debranching enzyme) regulates glycogen degradation as per the energy requirement of bacteria. Thus, the deletion of either HPr or GlgP or both prevents degradation of glycogen and these enzymes, involved in glycogen metabolism, are reported to act as important drug targets against M. tuberculosis. 57
Hypothetical carbohydrate phosphate isomerase (MAB_1574)
Carbohydrate phosphate isomerase represents the sugar isomerase enzymes ribose 5-phosphate isomerase B (RpiB), galactose isomerase subunit A (LacA) and galactose isomerase subunit B (LacB). Rpi plays an important role in bacterial growth 58 and is a homodimeric protein consisting of two isomerases RpiA and RpiB, which have different structures and active-site residues. Also, Rpi catalyzes the interconversion of D-ribose 5-phosphate and D-ribulose 5-phosphate in the nonoxidative branch of the pentose phosphate pathway. Carbohydrate phosphate isomerase has been identified as a putative target in Methicillin-resistant Staphylococcus aureus. 59
Acyl carrier protein (MAB_1878c)
Acyl carrier proteins (ACPs) are conserved α-helical bundle carrier proteins consisting of 70–100 amino acid residues. These proteins serve as essential cofactors in various metabolic pathways, including the biosynthesis of fatty acids, polyketides, phospholipids, and glycolipids. ACPs also act as a carrier molecule in reversible delivery of fatty acyl groups to a large class of ACP-dependent enzymes. 60 An extensive study on the fatty acid biosynthesis pathway from Escherichia coli 61 has suggested that this pathway could be an important target.
Two-component response regulatory protein (MAB_2627c)
A typical two-component system (TCS) consists of histidine kinase and a response regulator. TCSs are the key regulatory proteins in bacteria, which help them to respond aptly to environmental changes and control the expression of genes. Previous studies have demonstrated that TCSs are involved in virulence mechanisms in various pathogens like Streptococcus pneumonia, Vibrio cholerae, and Campylobacter jejuni.62–64 Furthermore, TCS proteins have been identified as potential targets in different bacteria, including Enterococcus faecium, S. aureus, and Salmonella enterica.65–67
4-Hydroxy-tetrahydrodipicolinate reductase, DapB (MAB_3096c)
The enzyme DapB consists of two domains. These comprise (1) an N-terminal domain made up of six-stranded sheet flanked by four helices, responsible for cofactor binding and (2) a C-terminal domain that plays a critical role in the tetramerization of the enzyme and also harbors the substrate-binding site.
68
This enzyme is involved in the catalysis of the bacterial biosynthetic pathway that generates meso-diaminopimelate and
Conserved hypothetical protein (MAB_3378c)
The protein MAB_3378c, in the present study, was found as a conserved hypothetical protein. However, the functional analysis indicated that it is a bacterial transferase hexapeptide repeat. Many bacterial transferases contain tandem repeats of [LIV]-G-X(4) hexapeptide and has been reported to form the tertiary structure of left-handed parallel beta-helix. 71 These transferase protein families include UDP N-acetylglucosamine acyltransferase (LpxA), galactoside acetyltransferase-like proteins, 72 and the gamma-class of carbonic anhydrases. 73 These proteins could be used as excellent drug targets.
Acetyl-CoA carboxylase biotin carboxyl carrier protein subunit (MAB_3541c)
Biotin acts as a catalyst in some carboxyl transfer reactions. Wherever required, it attaches covalently to a lysine residue through an amide bond. 74 This protein family is a component of acetyl coenzyme A carboxylase complex that is involved in the first step of the long-chain fatty acid synthesis. The transcarboxylase complex transfers the carboxyl group from the intermediate to acetyl-CoA forming malonyl-CoA 75 in the next step. The study on E. coli has demonstrated the validation of the biotin carboxylase as a novel target for antibacterial development. 76
UDP-glucose 4-epimerase GAlE1 (MAB_4003c)
The enzyme UDP-glucose 4-epimerase is known to catalyze the interconversion between UDP-galactose and UDP-glucose. The UDP-galactose aids in the synthesis of carbohydrate polymers, including bacterial virulence factors lipopolysaccharide and exopolysaccharide. 77 This enzyme is also a pivotal part of the Leloir pathway, which metabolizes galactose through glycolysis. 78 Previous studies have reported that the GalE is involved in pathogenicity in different species like C. jejuni 77 and Pasteurella multocida. 79
Succinate dehydrogenase, iron-sulfur subunit (MAB_4423)
Succinate dehydrogenase, also known as complex II, is a major respiratory enzyme that couples the oxidation of succinate to fumarate in the cytoplasm. This serves as a vital link between the tricarboxylic acid cycle and oxidative phosphorylation, and in the reduction of quinone to quinol in the membrane. It undergoes oxidative phosphorylation in the central carbon metabolism of the TCA. 80 Most of the mycobacterial genomes contain two annotated succinate dehydrogenases, designated as Sdh1 and Sdh2. Sdh1 is nonessential for growth, but Sdh2 is essential and generates the membrane potential under hypoxia. 81
30S ribosomal protein S18 and S6 (MAB_4897c and MAB_4899c)
Ribosomes are involved in the catalysis of mRNA-directed protein synthesis in all organisms. The small ribosomal subunit protein S18 is found to be involved in binding the aminoacyl/tRNA (transfer RNA) complex. 82 It also forms a heterodimer with protein S6 to the central domain of the 16S rRNA, where it helps stabilizing 30S subunit. Previous studies reported mycobacterial ribosomes as an important target for antitubercular drugs. 83
Single-stranded DNA-binding protein (MAB_4898c)
Single-stranded DNA-binding protein (SSB) is necessary for various DNA functions, including DNA replication, repair, and recombination. 84 SSBs are typically homotetramers consisting of four short peptides (150–180 amino acids) with regions of high conservation, particularly within the single-stranded DNA (ssDNA)-binding domain (1–110 amino acids). The short length and conserved nature of SSBs are ideal for drug targeting, as there is a reduced propensity for mutations causing resistance to arise. SSBs also possess a topography containing several cavities, which is ideal for binding small-molecule inhibitors. SSBs have been identified as potential targets in S. aureus. 85
Conclusions
The emergence of drug resistance among bacteria is a growing challenge for the treatment of infectious diseases. Exploring the novel targets in a pathogen could lead to the discovery of novel classes of antibacterial agents. The present study has elucidated a set of target candidates that can aid in designing effective broad-spectrum antimycobacterial agents against NTM. Proteins satisfying the criteria—core, pathogen-specific, essential, and druggable—are considered as broad-spectrum targets. The study has shortlisted 15 druggable candidates, among which 2 are virulence factors. The identified 15 candidate proteins are from different functional categories. Of 15 targets, a total of 13 targets are present in the cytoplasm, 1 in the extracellular region, and 1 is located in the cell membrane, and can be represented as drug/vaccine targets. These candidate proteins can be further explored for structural studies to design inhibitors.
Footnotes
Acknowledgments
The authors acknowledge the support of the Center for Bioinformatics, Pondicherry University (Pondicherry, India) and the Department of Biological Sciences, Sunway University (Selangor, Malaysia) for providing the computational facilities.
Disclosure Statement
No competing conflict or financial interests exist.
Funding Information
A.S. is thankful to the Department of Science and Technology, Government of India for providing DST-INSPIRE JRF fellowship. P.G. is grateful to the Department of Biotechnology, Government of India for providing DBT-BINC Junior Research Fellowship. J.P. is indebted to Indian Council of Medical Research (ICMR), Government of India for ICMR-SRF fellowship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.