
Abstract
Aim:
Genomic analysis of a methicillin-resistant Staphylococcus aureus (MRSA) strain cultured from a non-migratory seabird at Fernando de Noronha Archipelago (Brazilian oceanic islands) was carried out to investigate the potential origin of MRSA genetic determinants in an ecological setting with minimal or absent antimicrobial selective pressure, and minimal interaction with humans and domestic animals.
Results:
The study determined mecA gene homology and the phylogenetic relatedness with mecA described in Staphylococcus sciuri, which was the major Staphylococcus spp. cultured from the birds. Our findings corroborate in silico assumptions that the mecA gene in MRSA strains clinically relevant for humans and animals originates from S. sciuri ancestors.
Conclusion:
Coagulase-negative staphylococci seem to be natural reservoirs of methicillin-resistant genes to S. aureus, even in environments with very low antimicrobial selection pressure.
Introduction
Antimicrobial resistance in pathogenic bacteria represents a major threat to the global public health. Due to the alarming rates of increase in antimicrobial resistance, a public health crisis has been deemed by the United Nations Interagency Coordination Group on Antimicrobial Resistance.1,2 Methicillin-resistant Staphylococcus aureus (MRSA) are S. aureus harboring staphylococcal cassette chromosome mec, a mobile element that encodes an alternative penicillin-binding-protein (PBP2a) conferring resistance to virtually all β-lactam drugs.
MRSA has been commonly associated with hospital-acquired infections (HA-MRSA). In the past few decades, however, community-associated strains (CA-MRSA) have also emerged as important pathogens in urban and rural settings. 3 More recently, genomic studies suggested that asymptomatic livestock, such as swine 4 and cattle, 5 could be reservoirs of certain MRSA lineages, also named livestock-associated MRSA, which cause occupational infections in humans. However, there are very limited data on MRSA in wildlife ecological settings and there is paucity of information on the evolution and epidemiology of MRSA strains and the role of wild fauna as potential sources of MRSA to humans and domestic animals.
Comparative investigations of Staphylococcus genomic sequences have shown that staphylococci from animals and humans share a large number of antimicrobial resistance determinants. 6 Both S. aureus7–9 and species other than S. aureus recovered from domestic and wildlife animals have been reported to carry mecA9–11 or its variant mecC.12,13
Although there is a consensus that both domestic and wildlife species are potential MRSA reservoirs to humans, the significance of wildlife species as mecA reservoirs is difficult to discern, since most studies include wildlife host species in urban dwelling ecosystems such as parrots, parakeet, and gouldian finch.14,15
We believe that information on native methicillin-resistant mechanisms in S. aureus from environments with none or little antimicrobial selective pressure could shed light on the understanding of the emergence and dissemination of methicillin resistance. In this study, we reported the antimicrobial resistance patterns of Staphylococcus spp. cultured from non-migratory seabirds inhabiting Fernando de Noronha Archipelago, a secluded Brazilian island located in the Atlantic Ocean. We also performed in-depth whole genome sequencing of an MRSA strain to investigate the genetic context of methicillin resistance.
Methods
The study proposal was submitted and approved by the Ethics Committee on Animal Use of Research and licensed by Biodiversity Information System (SISBIO) from Brazilian Chico Mendes Institute for Biodiversity Conservation (ICMBio). All methods, including animal handling and sampling procedures, were performed in accordance with SISBIO/ICMBio approved regulations. The samples were collected from endangered seabird populations in a non-tourist area in Fernando de Noronha Archipelago, a group of 21 oceanic islands and islets about 354 km from the Brazilian mainland, covering a total area of 26 km2 (Fig. 1).
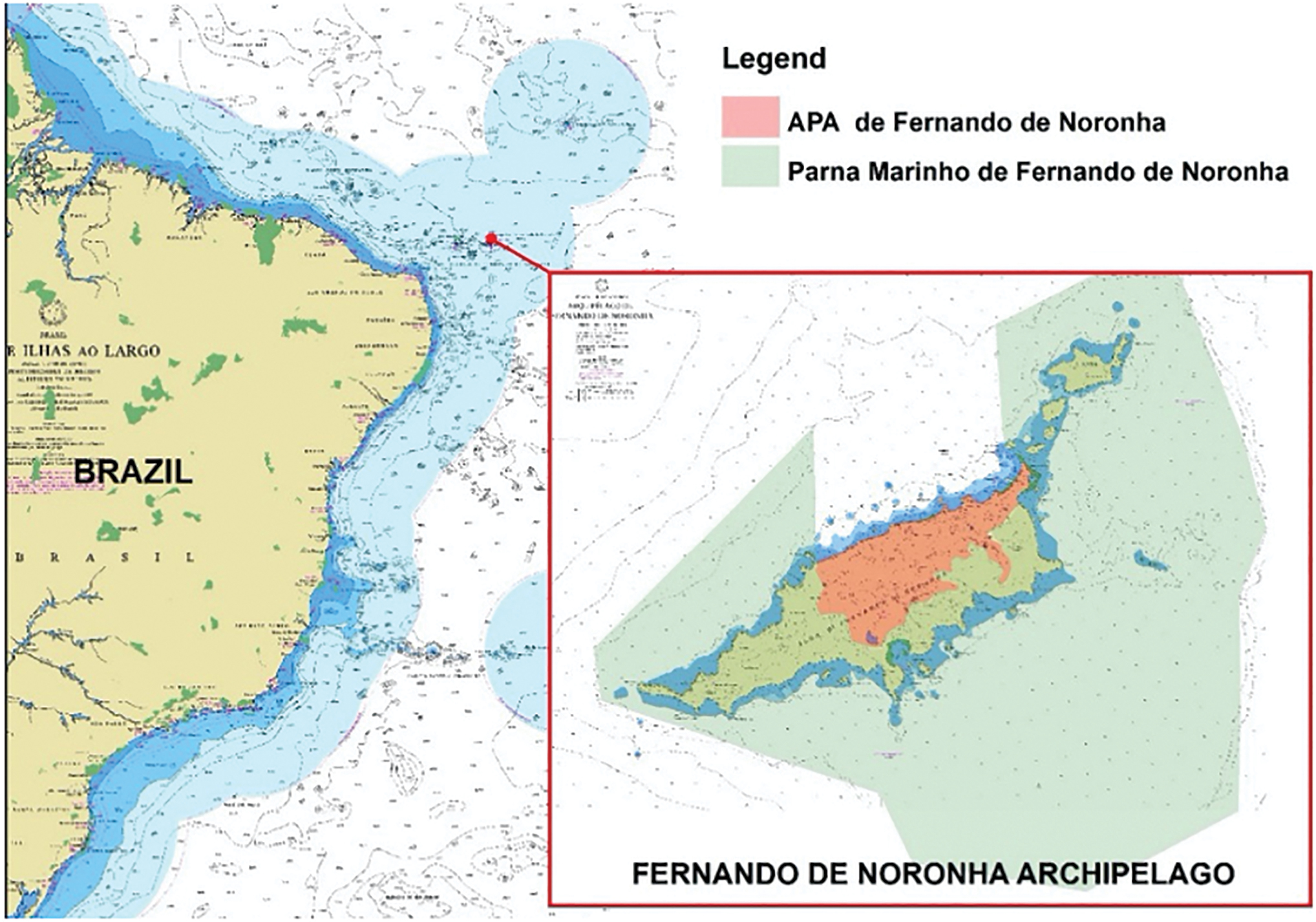
Study area. Fernando de Noronha Archipelago, Brazil. The two federal protected areas are featured on the map in different colors. Map designed by Leandro Zago (ICMBio). APA, Environmental Protection Area; Parna, Marine National Park of Fernando de Noronha. Color images are available online.
Adult birds were sampled in their nests, using climbing techniques to reach the mountain sides. Researchers used personal protective equipment to avoid contamination between birds and humans (Supplementary Fig. S1). After manual restraint, cloacal and tracheal swabs were collected from 16 White-tailed tropicbirds (Phaethon lepturus, order Phaethontiformes) (n = 16) and two Audubon's shearwater birds (Puffinus lherminieri, order Procellariiformes).
Swab samples were kept in Eagle's minimal essential medium until laboratory analysis. Microbiological isolation was performed by using blood agar (Oxoid, UK) enriched with 5% sheep's blood and mannitol-salt agar (Himedia, India). Isolates showing typical and atypical morphological characteristics of Staphylococcus genus were further confirmed by Gram staining, catalase, and oxidase. Staphylococci were tested for coagulase in tubes and screened for methicillin resistance by using Muller-Hinton (Himedia) agar supplemented with oxacillin (7 mg/L).
Species identification and antimicrobial susceptibility testing were performed by using a biochemical panel (PC Combo 33; Siemens Healthcare, USA) by means of a semi-automated system (Autoscan4; Siemens Healthcare) using a commercial software (LabPro; Siemens Healthcare). The minimum inhibitory concentrations and cut-off values were determined for ampicillin (Amp); amoxicillin/clavulanate K (Amc); ampicillin/sulbactam (Ams); ceftriaxone (Cax); clindamycin (Cli); ciprofloxacin (Cip); daptomycin (Dap); erythromycin (Ery); gentamicin (Gen); levofloxacin (Lvx); linezolid (Lzd); moxifloxacin (Mox); nitrofurantoin (Nit); oxacillin (Oxa); penicillin (Pen); rifampin (Rif); quinupristin-dalfopristin (Q/D); sulfisoxazole/trimetropim (Sxt); tetracycline (Tet); and vancomycin (Van). Staphylococcus aureus ATCC 25923 was used as a control, and results were interpreted following the criteria proposed by the Clinical and Laboratory Standards Institute (2018). 16
Staphylococcus spp. were further confirmed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) (Vitek MS; bioMérieux, Mercy l'Etoile, France). Briefly, bacteria were cultivated in trypticase soy agar (Himedia) for 24 hours at 37°C. Colonies were then transferred to a 48-well standard steel plate and covered with a matrix solution of α-cyano-4hydroxycinnamic acid. The plates were dried at room temperature before MALDI-TOF analysis. The obtained mass spectra were compared with onboard database for species identification.
DNA extraction was performed after sample incubation at 37°C for 24 hours in trypticase soy agar (Himedia). Bacterial scrapings were transferred to microtubes with 1 mL utrapure water and centrifuged at 5,670 g for 10 minutes. The pellet was resuspended and DNA was extracted by using a commercial kit (PowerSoil DNA Isolation; Qiagen, USA), following the manufacturer's recommendations. DNA quantification was performed by using a fluorometer (Qubit 3.0; Invitrogen, USA).
DNA library was prepared by using the Nextera XT Sample Prep Kit (Illumina, Inc., USA), and DNA fragments were purified by Agencourt AMPure XP reagent (Beckman Coulter, USA). The library was validated by using a capillary electrophoresis system (Fragment Analyzer; Agilent Technologies, USA). Paired-end sequencing was performed on Miseq by using a 300 bp V3 kit (Illumina, Inc.) according to the manufacturer's recommendations. The quality of the reads was assessed by using FastQC 0.11.7 (Ref. 17 ). Illumina adapters and low-quality reads (Phred <25) were filtered by using Trimmomatic-0.38 (Ref. 18 ).
Genome assembly and annotation were performed by using Spades on Pathosystems Resource Integration Center 19 and Rapid Annotations using Subsystems Technology, 20 respectively. Epidemiologic information was obtained from the Center for Genomic Epidemiology by means of spa typing (spaTyper-1.0), 21 Multilocus sequence typing (MLST) (MLST-2.0), 22 antimicrobial resistance genes (ResFinder-3.1), 23 virulence genes (VirulenceFinder-2.0), 24 and the predicted pathogenicity toward human hosts (PathogenFinder). 25
Analyses of specific genes were performed by using Artemis Comparison Tool 26 and SeaView v4. 27 Phylogenetic analysis was performed by using maximum likelihood with 1,000 repetitions of bootstrap by means of RAxML-HPC BlackBox 8.2.10 on CIPRES Science Gateway. 28
Results
Out of the 36 samples collected from 18 birds, 30 staphylococci isolates were recovered. A total of 15 birds (75%) were found to be positive for Staphylococcus. Six birds were positive for cloacal swabs only, three for oral swabs only, and six for both types of samples. Five staphylococcal species were detected and confirmed by MALDI-TOF (Supplementary Table S1): Staphylococcus sciuri (22 isolates, 73.3%), Staphylococcus intermedius (4; 13.3%), Staphylococcus saprophyticus (2; 6.7%), S. aureus (1; 3.3%), and Staphylococcus haemolyticus (1; 3.3%). Five birds had both cloacal and tracheal swabs that were positive for S. sciuri. Co-infection with different staphylococcal species within the same bird was found in cloacal swabs of two birds (S. sciuri/S. saprophyticus and S. sciuri/S. haemolyticus) and a tracheal swab of one bird (S. sciuri/S. aureus).
The results of the antimicrobial susceptibility test are shown in Table 1. All isolates were resistant to Amp and Cax, whereas only the S. aureus was resistant to Q/D and Tet. All isolates were susceptible to Ams, Cip, Nit, Gen, Lvx, Lzd, Mox, Sxt, and Van.
Minimum Inhibitory Concentration of Antimicrobial Drugs Against 30 Staphylococci Recovered from White-Tailed Tropicbird (Phaethon lepturus) and Audubon's Shearwater Birds (Puffinus lherminieri) at Fernando de Noronha Archipelago, Brazil
Antimicrobials: Amp, Amc, Ams, Cax, Cli, Cip, Dap, Ery, Gen, Lvx, Lzd, Mox, Nit, Oxa, Pen, Rif, Q/D, Sxt, Tet, and Van.
Amc, amoxicillin/clavulanate K; Amp, ampicillin; Ams, ampicillin/sulbactam; Cax, ceftriaxone; Cip, ciprofloxacin; Cli, clindamicyn; Dap, daptomycin; Ery, erythromycin; Gen, gentamicin; Lvx, levofloxacin; Lzd, linezolid; Mox, moxifloxacin; Nit, nitrofurantoin; Oxa, oxacillin; Pen, penicillin; Q/D, quinupristin/dalfopristin; Rif, rifampin; Sxt, sulfisoxazole/trimetropim; Tet, tetracycline; Van, vancomycin.
All 25 coagulase-negative staphylococci (CoNS) isolates phenotypically resistant to methicillin were β-lactamase producers; all non-β-lactamase producers were S. sciuri isolates recovered from a single bird (n = 4). From the six different detected phenotypes, Amp–Amc–Cax–Oxa (72.41%) was the most frequent resistance profile. Seven isolates (24.15%) were resistant to three or more different classes of drugs and were considered multidrug resistant (Table 2).
Antimicrobial Resistance Profiles of Staphylococcus spp. Recovered from White-Tailed Tropicbird (Phaethon lepturus) and Audubon's Shearwater Birds (Puffinus lherminieri) at Fernando de Noronha Archipelago, Brazil
Antimicrobial drugs: Amp; Amc; Cax; Cli; Dap; Ery; Oxa; Pen; Rif; Q/D; and Tet.
Multidrug-resistant pattern.
Multidrug-resistant pattern from mecA-producing S. aureus.
The single S. aureus isolate found in this study was resistant to eight antimicrobials, including Cax, Oxa, Pen, Q/D, and Tet, presenting a multidrug resistance pattern: Amp–Cax–Cli–Dap–Ery–Oxa–Pen–Rif–Q/D–Tet (Table 2).
Whole genome sequencing of this isolate generated 1,358,129 reads with ∼291 × of coverage. After sequence trimming, 891,545 surviving reads (191 × coverage) were used for the assembly of a 2,914,355 bp draft genome containing 93 contigs with 32.7% CG content. Genome annotation showed 4,734 coding sequences and 98 RNAs in 327 subsystem categories.
Based on in silico analyses, the strain was identified as sequence type 7 (ST7) and spa type t091. This isolate harbored genes conferring resistance against drugs from four different classes: fosfomycin (fosD); MLS-macrolide, lincosamide, and streptogramin (lnuA; salA); tetracycline (tetK); and β-lactam (mecA). However, neither mecR1 nor mecI (mec regulatory sequences) was found. Different virulence factors were also detected (Table 3), including toxins (hlgA, hlgB, hlgC, lukD, and lukE), exoenzymes (aur, splA, splB, and splE), and genes encoding immune-modulating proteins—hostimm (sak and scn). By means of PathogenFinder, this strain was predicted to be pathogenic to humans with 93.6% probability (Supplementary Fig. S2).
Genomic Features Including spa Type, Sequence Type, Resistance, and Virulence Genes Identified by Whole Genome Sequencing of a Methicillin-Resistant Staphylococcus aureus Strain Recovered from a Non-migratory White-Tailed Tropicbird at Fernando de Noronha Archipelago, Brazil
Genes encoding immune-modulating proteins.
MLS, macrolide, lincosamide, and streptogramin; MLST, multilocus sequence typing; ST7, sequence type 7.
Phylogenetic analyses (Fig. 2) revealed higher homology of the MRSA ST7 mecA gene to the PBP2 protein-encoding gene from S. sciuri strains (accessions: Y13094; NZ_CP020377.1; KX774480.1; QXVD01000001.1; Y13052.1) than to the mecA gene from unrelated S. aureus (accessions: BX571856; KT780704; HE579063; FN433596; NG_047948; NZ_CP029030; NZ_CP018768; NZ_CP020019; AP019751; NC_017349; NZ_CP028165), Staphylococcus vitulinus (accession: AB546780.1), and Staphylococcus fleurettii (accessions: UHDL01000001.1; AB546267.1). Table 4 and Supplementary Figure S3 present the homology information at the protein level.
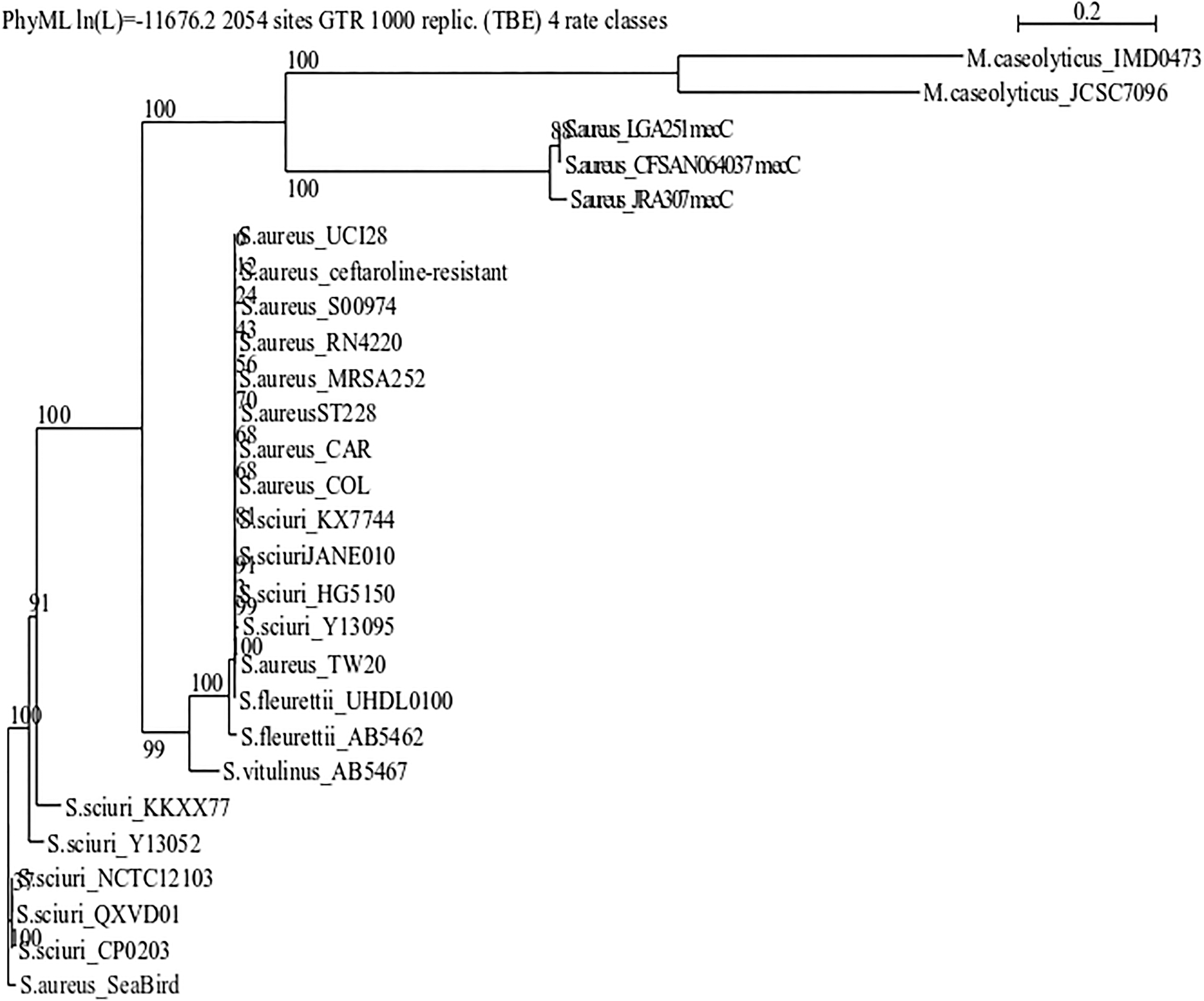
Phylogenetic analysis of the mec gene harbored by different Staphylococcus strains: Staphylococcus aureus ST7 from White-tailed tropicbird (Phaethon lepturus) at Fernando de Noronha Archipelago, Brazil (this study), Staphylococcus sciuri (accessions: Y13094; KX774481.1; NZ_JANE01000001.1; HG515014.1; NZ_CP020377.1; KX774480.1; QXVD01000001.1; Y13095.1; Y13052.1), mecA-carrying S. aureus (accessions: BX571856; KT780704; HE579063; FN433596; NG_047948; NZ_CP029030; NZ_CP018768; NZ_CP020019), mecC-carrying S. aureus (accessions: AP019751; NC_017349; NZ_CP028165), Staphylococcus vitulinus (accession: AB546780.1), Staphylococcus fleurettii (accessions: UHDL01000001.1; AB546267.1), and Macrococcus caseolyticus (accessions: NG_054960; NG_047953). The bootstrap values support the clades in the complete tree building. ST7, sequence type 7.
Homology between mecA-Encoded Amino Acid Sequences from a Staphylococcus aureus Sequence Type 7 Strain Recovered from a White-Tailed Tropicbird Seabird (Phaethon lepturus) at Fernando de Noronha Archipelago, Brazil (667 aa; S. aureus Seabird, This Study), a Reference S. aureus Strain (669 aa; S. aureus COL; CP000046.1), and a Reference S. sciuri Strain (667 aa; S. sciuri NCTC12103; Y13094)
Bold letters mean different amino acid at the same locus.
NCTC, National Collection of Type Cultures.
Discussion
Previous studies have reported the occurrence of different Staphylococcus spp. in seabirds in Brazil 29 and in other countries. 14 Staphylococcal species such as S. aureus, S. sciuri, and S. xylosus have been recovered from wild birds in illegal trade and wild birds of prey.30,31 however, Staphylococcus spp. occurrence in wild birds under very low or no antimicrobial selection pressure is very scarce.
High antimicrobial resistance rates have been reported in bacteria from Brazilian seabirds, mainly against rifampicin, erythromycin, ciprofloxacin, norfloxacin, and tetracycline. 32 However, resistance to β-lactams in Staphylococcus has been reported only in migratory corvus, 12 water birds, 33 birds of prey, 9 and marine mammals. 7 A more recent study on migratory gulls in Europe 34 reported staphylococci harboring genes that confer resistance to ampicillin, ampicillin/sulbactam, and colistin. Our findings suggest that antimicrobial resistance in staphylococci from non-migratory bird species in this remote island is mainly related to β-lactams. Exposure to β-lactam drugs determined by anthropogenic causes has been considered a key driver leading to methicillin resistance through PBPs mechanisms. 35 Although it is hard to predict the degree of contact of those species with humans, the fact remains that they are non-migratory birds in an archipelago with strict conservations laws, suggesting a singular situation with very low antimicrobial resistance pressure, at least significantly lower than in other areas. This is noteworthy considering the peculiar life cycle of the investigated seabird species, which use only pelagic areas for feeding and oceanic islands for breeding. 36 Besides, visitations of tourists or even inhabitants are not allowed in three of the four islands where the birds were sampled. Therefore, both species have virtually no contact with humans or domestic animals, and no antimicrobial selection pressure on those populations is expected.
Staphylococcus spp. identification by means of biochemical tests is time-consuming and can lead to misidentification, because capacity to metabolize some sugars can vary within a given species. 37 These limitations have been overcome by using MALDI-TOF mass spectrometry, which has been consolidated as a gold standard method in bacterial identification. 38
Different MRSA sequence types isolated from wildlife, including birds, have been shown to harbor several resistance genes (blaZ, mecC, and tetK) and virulence genes (lukD, lukE, and seh).13,31 ST7 is not among the most reported STs in staphylococci, but it is usually associated with commensal Methicillin Susceptible Staphylococcus aureus strains in animals (pets, food animals, and wildlife) and humans.39–42 Interestingly, the repertoire of virulence factors carried by the MRSA ST7 strain reported in this study was highly correlated with those found in Staphylococcus strains associated with human infections. Although mecA is not related to increased virulence, 43 it confers competitive advantages to survive in different environments. 44
The mecA gene from S. sciuri has been suggested to be the precursor of the mecA gene found in clinically relevant S. aureus currently associated with infections in humans. 35 Our findings clearly demonstrate a high homology between the mecA gene harbored by the MRSA strain and the mecA found in S. sciuri, as well as high homology at the protein sequence level, thus corroborating the hypothesis that S. sciuri is the ancestral reservoir of mecA gene. This is also supported by the frequent presence of S. sciuri spp. in the samples taken from the same population.
Although different mec nucleotide sequences have been described in both coagulase-positive Staphylococcus and CoNS strains,45,46 this is the first study to report the homology of mecA found in S. aureus isolated from a low antimicrobial selective pressure environment.
White-tailed tropicbird (P. lepturus) and Audubon's shearwater (P. lherminieri) are considered threatened species in Brazil. Fernando de Noronha archipelago is the exclusive breeding site for the Audubon's shearwater, and one of the only two breeding sites for the White-tailed tropcbird. 36 Therefore, both are particularly vulnerable to invasive exotic species, disease introduction, and habitat destruction. A continuous monitoring of population health is necessary to follow new ecological or disease distributions, predict population trends, and evaluate the overall health status of the marine ecosystem. 36
In summary, studying antimicrobial resistance in wildlife under low antimicrobial pressure represents a unique opportunity to understand the origin and spread of resistance determinants among microbial pathogens of veterinary and public health significance. The present findings support the hypothesis that S. sciuri is a major reservoir of mecA determinant to S. aureus spp. The genome sequence made available by this study can support further comparative genomic investigations addressing evolutionary and epidemiological aspects of β-lactam resistance.
Footnotes
Authors' Contributions
C.J.B.O., P.E.N.G., and M.M.S.S. constructed the hypothesis; T.F.R. and P.P.S. performed the samplings; P.P.S., C.M.C.G.L., and M.M.S.S. performed the microbiological culture and species identification; N.M.V.S. and M.M.S.S. sequenced the strain and performed the bioinformatics analyses; and W.A.G., C.J.B.O., P.E.N.G., and T.F.R. wrote the article. All authors reviewed the article.
Disclosure Statement
The authors declare no conflict of interest.
Funding Information
This study was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES, Finance Code 001), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp), and Financiadora de Estudos e Projetos (FINEP). The authors thank ICMBio/NGI Noronha, ICMBio/CEMAVE, Fire Department, PARNAMAR, APA, and Administration of Fernando de Noronha for operational support for this research.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.