
Abstract
Antibiotic resistance genes (ARGs) have become recognized contaminants and pose a high public health risk. The animal gut microbiota is a reservoir of ARGs, but the knowledge of the origin and dissemination of ARGs remains unclear. In this study, we provide a comprehensive profile of ARGs and mobile genetic elements in the gut microbiota from 30 bovines to study the impact of modern antibiotics on resistance. A total of 42 ARG types were detected by annotating the metagenomic sequencing data from Comprehensive Antibiotic Resistance Database (CARD). We found that the diversity and abundance of ARGs in individual yaks were significantly lower than those in dairy and beef cattle (p < 0.0001). The results of heat map and single nucleotide polymorphism clustering suggest that ARGs from dairy and beef cattle are more similar, whereas those from yaks cluster separately. The long-term use of antibiotics may contribute to this difference, suggesting that antibiotic consumption is the main cause of ARG prevalence. Furthermore, abundant insertions were also found in this study, signifying a strong potential for horizontal transfer of ARGs among microbes, especially pathogens.
Introduction
The emergence and widespread presence of antibiotic resistance genes (ARGs) have become growing global public health issues.1,2 Moreover, the accelerating accumulation and evolution of ARGs among multidrug-resistant (MDR) pathogens pose a potential threat to humans and animals. 3
The abuse and overuse of antibiotics in animals suggest that hospitals are no longer the only source of drug-resistant bacterial dissemination. 4 Approximately 0.2 million tons of antibiotics are produced and consumed annually in China and are used in the livestock industry to treat clinical bacterial infections (therapy), prevent clinical infections (prophylaxis), or for nutritional purposes (growth promotion).4–6 The long-term use of antimicrobials has created a sustained selection pressure for resistant organisms, which will promote the spread of ARGs between symbiotic bacteria and pathogens. 7 Their capacity to engage in horizontal gene transfer (HGT) through mobile genetic elements (MGEs), such as insertion sequence elements (ISs), transposons, and plasmids, is responsible for the common existence of ARGs and the spread of MDR bacteria in modern environments. 8 Fournier et al. 9 reported that the MDR Acinetobacter baumannii strain AYE isolated in France contains 45 resistance genes in its chromosomal resistance gene island, most of which were acquired from the genera Pseudomonas, Salmonella, and Escherichia, whereas bacteria isolated from the pre-antibiotic era and wild animals in remote areas with no history of antibiotic exposure rarely carry ARGs.9–11 This situation indicates that there is a high risk that we will regress to an era where antibiotics are no longer effective and may contribute to the increased emergence of antibiotic-resistant pathogens.
The gut is inhabited by an abundant variety of microbial species. This day microbiota, which has been considered the second genome of a given organism, can mediate profound effects on human and animal nutrition, metabolism and physiological function. However, more attention must be paid to the other role that the gut microbiota plays as a reservoir of known and novel ARGs, which could be shared between resistant bacteria and the resident microbiota.5,12 Large numbers of ARGs for different types of antibiotics have already been identified by PCR, qPCR, and microarray analysis.13–15 At present, functional metagenomic is a powerful tool that enables the characterization and discovery of ARGs by high-throughput sequencing (HTS) when annotated in suitable databases, and plentiful ARGs have been identified in gut and environmental microorganisms.16–18 The analysis of metagenomic data has enabled a broad view for ARG identification.
The objectives of the present study were (1) to examine the diversity and abundance of ARGs and MGEs in the bovine gut microbiota and (2) to reveal the correlations between modern antibiotics and ARG prevalence.
Materials and Methods
Fecal sample collection
Our previous study was conducted with approval from the Ethics Committee of Animal Experiments at the Institute of Husbandry and Pharmaceutical Sciences of CAAS in Lanzhou, China. Samples for metagenomes in this study were obtained from a total of 6 farms in the Gansu province of China, including dairy cattle (Zhangye n = 5, Jinchang n = 5), beef cattle (Zhangye n = 5, Linxia n = 5), and yak (Tianzhu n = 5, Gannan n = 5). Each bovine was selected with similar weight and age on the same farm to investigate the history of antibiotic exposure. Fresh fecal samples of ∼100 g were collected at the rectum from each bovine and were immediately frozen in liquid nitrogen. The basic information for the 30 samples in this study is given in Table 1. All samples were kept on dry ice during transportation and were stored at −80°C before DNA isolation.
The Basic Information of the Fecal Samples
DNA extraction and metagenome sequencing
Total metagenomic DNA extraction from the fecal samples was performed using the cetyltrimethylammonium bromide method. The degree of degradation and the potential contamination of the extracted DNA were monitored on 1% agarose gels, and the purity was checked using a NanoPhotometer® spectrophotometer (IMPLEN, CA). DNA concentration was measured using a Qubit® dsDNA Assay Kit in a Qubit 2.0 Fluorometer (Life Technologies, CA). Approximately 5 μg of each DNA sample was used for shotgun library construction. Sequencing was then performed on the Illumina HiSeq X Ten platform, and 150-bp paired-end reads were generated. Adaptor contamination, low-quality reads, and host contaminating reads were removed from the raw sequencing reads sets. To ensure the reliability of the results, the size of the output data for each sample was >12 Gb, and the proportion of high-quality reads among all raw reads from each sample was no less than 95%.
Metagenome assembly and ORF prediction
After quality control, we assembled high-quality clean reads into Scaftigs for each of the samples using SOAPdenovo2 19 with a Kmer of 55.20–23 For each sample, the clean data were mapped to Scaftigs using SoapAligner. 22 Assembled Scaftigs longer than 500 bp were used for downstream gene prediction.21,22,24,25
The open reading frames (ORFs) within the Scaftigs (≥500 bp) used MetaGeneMark.24,26 To identify the nonredundant Unigenes, all the ORFs should be filtered using CD-HIT with a minimum similarity of identity of 95% and a coverage of 90%, and the longest gene sequence was selected as the standard. 27 The coverage of each gene per sample was quantified by mapping the metagenomic reads to the ORF.28,29
Relative abundance analysis
The abundance of a gene was calculated by counting the number of reads that align to the gene normalized by the gene length and the total number of reads aligned to any contig.
30
For each gene, r is the number of read pairs, L is the length of the corresponding gene, and G is the relative abundance. The k and i are the serial number of sample and read pairs. The relative abundance of different genes was calculated using the following formula:
Taxonomic assignments
The taxonomic affiliations of the microorganisms in the cattle gut were analyzed with MEGAN 31 using the lowest common ancestor (LCA) algorithm (LCA parameters: mini-score 35, top percentage 10%). Before applying this program, the Unigenes sequences from each sample were compared against the NCBI NR database (bacteria/archaea genome database, version: 2015-12-04) using DIAMOND under the default parameters (BLASTp, E-value ≤1e-5). 32
Identification of ARGs and MGEs
A commonly accepted ARG annotation tool together with a core set of 7,828 antibiotic resistance proteins was downloaded from the Comprehensive Antibiotic Resistance Database (CARD, version 1.1). 33 All nonredundant genes cataloged in each set of metagenomic sequencing data were aligned with the resistance proteins using BLASTx with an E-value threshold of 1e-5.17,34 A gene was annotated as an ARG-like sequence if the best BLAST hit (blastx) had a sequence identity of higher than the CARD requirement.
In this study, an MGE database was constructed based on 472 ISs, which were downloaded from the ISfinder database. All the metagenomic sequencing data were aligned against the insertion databases to investigate the prevalence of these two types of MGEs. A sequence was annotated as an MGE sequence if the best BLAST hit (blastn) had a nucleotide sequence identity higher than 90% over an alignment length of at least 50 bp. 35
Statistical analysis
Statistical analysis was performed using SPSS 19.0 (SPSS, Inc., Chicago, IL). Correlation analysis was conducted using the Pearson test, and p < 0.05 was considered statistically significant. The averages and standard deviations of all data were processed using Microsoft Excel 2007 (Microsoft Corp., Redmond, WA).
Results
Identification and taxonomy of ARGs
The animal gut microbiota has become a large and flexible gene reservoir for ARGs. To assess the potential impact for the ARG reservoir by antibiotics, we constructed a metagenomic library from yaks and dairy and beef cattle. A total of 403.20 Gb of high-quality data with an average of 13.44 Gb per individual was generated by the deep sequencing of 30 fecal DNA samples, which allowed us to identify 4.1 million nonredundant genes (Supplementary Table S1). Next, we compared the nonredundant gene sets with the CARD to search for ARG-like genes. After manually checking and removing the low-quality sequences in the original CARD, we identified a total of 4,739 ARGs that accounted for 1.16‰ of the total nonredundant gene set.
To study the dominant species carrying ARGs and the ARG types, we assigned the set of 4.1 million nonredundant genes and the 4793 ARGs according to the NCBI taxonomy guidelines with the assistance of the MEGAN classification software system. The differences in the gut genes set and the ARGs were observed at the phylum level, which were 31% versus 68%, 40% versus 21%, and 4% versus 4% of Firmicutes, Bacteroidetes, and Proteobacteria, respectively (Supplementary Fig. S1). These incongruous results suggest that ARGs are more prone to occur in Firmicutes but are less prone to exist in Bacteroidetes in bovine gut microbiota. More interestingly, each bacterial species had their own resistance strategy. Ribosomal protection proteins, RND transporters, and beta-lactamase class A proteins were the main resistance mechanisms of Firmicutes, Proteobacteria, and Bacteroidetes, respectively (Supplementary Fig. S2).
Diversity and distribution of ARGs
All the 4739 ARGs were grouped into 42 distinct ARG types (10 were MDR and 32 were single drug class resistance gene types), which could confer resistance to 11 different classes of antimicrobials. To study the distribution of ARG types, we mapped all the ARGs to each individual. In total, 42, 39, and 21 ARG types were identified in dairy cattle, beef cattle, and yaks, respectively (Supplementary Fig. S3). Of note, all the 21 ARG types found in the yak were also found in the dairy and beef cattle groups. Among the 30 individuals, one dairy cattle, DZY04, had the highest number of ARG types (21), whereas yaks YTZ02 and YTZ04 exhibited the lowest numbers of ARG types (3) (Supplementary Fig. S3).
For the 4,739 ARGs, the dairy cattle group harbored the most, 2,030 ARGs, followed by the beef cattle at 1,642 and yaks at 1121. To eliminate the bias caused by the basic data, we computed the gene number per gigabase pairs (Gb) in each individual (Supplementary Table S2). We found that dairy (15.19 ± 1.2 per Gb) and beef cattle (12.15 ± 2.43 per Gb) exhibited more complete ARGs than yaks (8.34 ± 0.58 per Gb) (p < 0.01).
Of the 42 ARG types, 2, 13, and 15 ARG types were found in every individual in the yak, dairy, and beef cattle groups, respectively. Furthermore, 13 ARG types were commonly found in each dairy and beef cattle sample, including tet32, tet40, tetO, tetPa, tetQ, tetW, tetX, ant6ia, ermF, bl2e_cfxA, mefA, mphB, and bacA. Among them, the most prevalent ARGs were bl2e_cfxA and bacA, which were found in all 30 individuals and conferred resistance to cephalosporin and bacitracin, respectively (Fig. 1 and Supplementary Fig. S3). Of interest, the most diverse ARGs, such as tetracycline resistance genes (TER), aminoglycoside resistance genes, MLS resistance genes, sulfonamide resistance genes, and macrolide resistance genes, were widely found in the dairy and beef cattle individuals but were hardly found in yak.
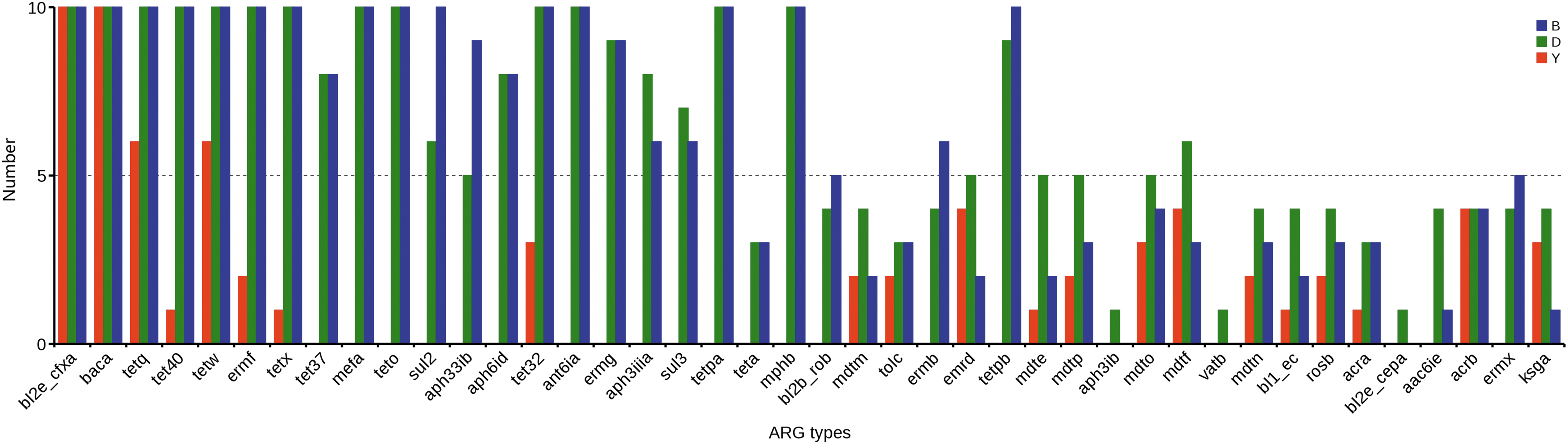
The distributions of 42 ARG types in different individuals. The x-axis is the name of resistance gene, and y-axis is the number of resistance gene in different sample. B = beef cattle; D = dairy cow; Y = yak. ARG, antibiotic resistance gene.
Relative abundance of ARGs
To investigate the abundance discrepancies of ARGs among yak, dairy, and beef cattle, we compared the relative abundance levels of all ARGs. Supplementary Table S3 summarizes the relative abundance levels of ARGs in each individual, which ranged from 1.18E-04 ppm (YTZ03) to 1.13E-03 ppm (DZY01). In general, environments that use antibiotics have the greatest abundance of ARGs. Compared with the ARG abundance in the yak (1.32E-03 ppm) group, the abundance levels of ARGs in both the dairy (5.60E-03 ppm) and beef cattle (5.80E-03 ppm) were significantly higher (p < 0.0001) (Fig. 2A and Supplementary Table S3). However, there was no significant difference in abundance levels between the dairy and beef cattle groups, indicating that ARGs were enriched in the gut microbiota in the host species that commonly used antibiotics. Furthermore, we also found that the abundance levels of ARGs in the Zhangye farms were significantly higher (p < 0.0001) than the other areas, at 3.62E-03 ppm versus 1.98E-03 ppm and 4.07E-03 ppm versus 1.73E-03 ppm in the dairy and beef cattle, respectively (Fig. 2B and Supplementary Table S3).

Boxplot showing the relative abundance levels of the total ARGs in each group. The individuals are shown as dots on the left. The asterisks on the top indicate p < 0.0001 (Mann–Whitney test).
We also analyzed the relative abundance distributions of the classes of ARGs across the yak, dairy, and beef cattle (Fig. 3, Supplementary Fig. S4, and Supplementary Table S3). Obviously, genes providing resistance to tetracycline (4.63E-03 ppm) were the most common type of ARGs, followed by bacitracin (4.11E-03 ppm) and beta-lactam (2.42E-03 ppm) resistance genes.
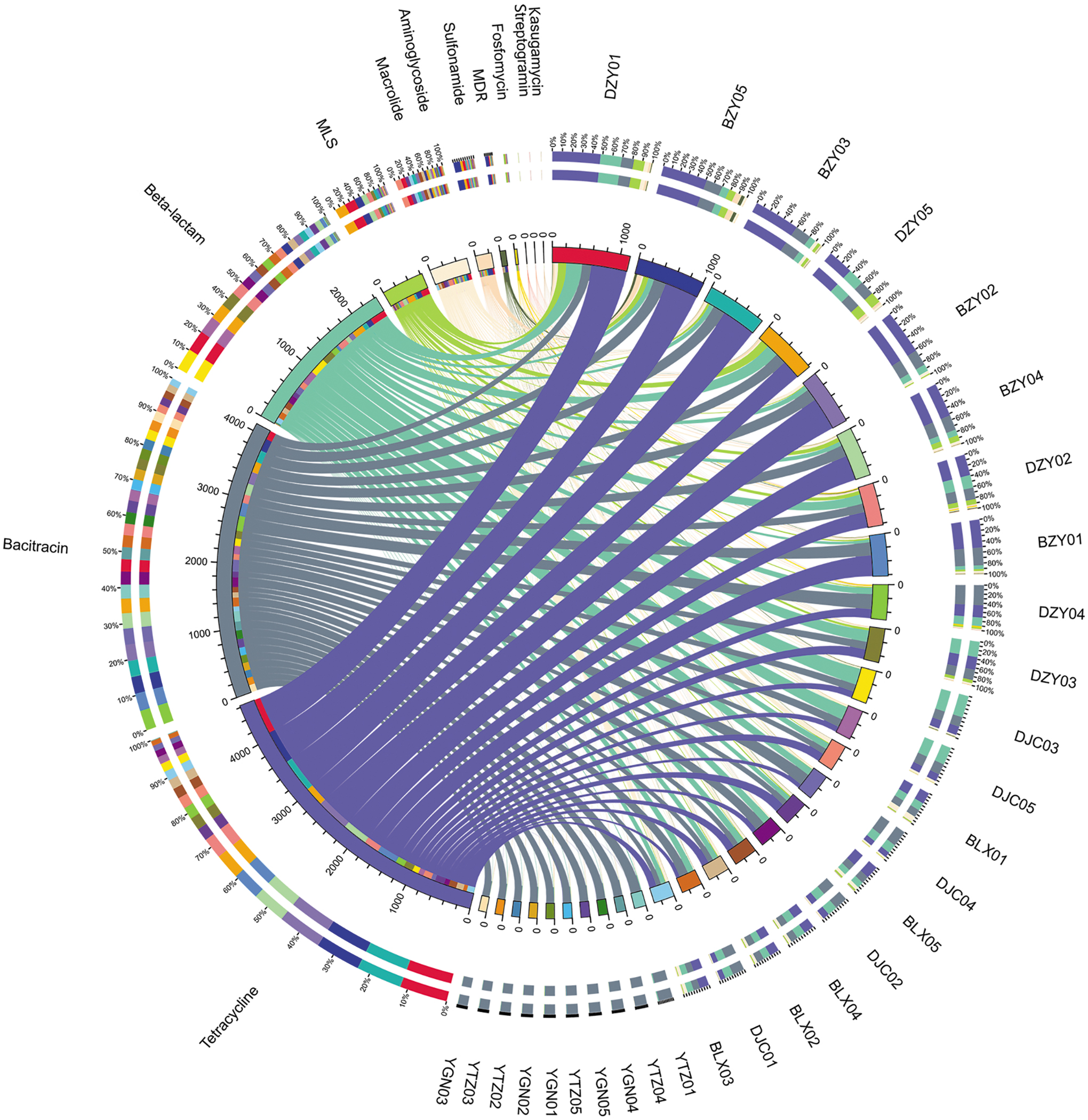
Overview map of the distributions of each ARG type in 30 individuals. The lengths of the bars on the outer and inner rings represent the percentages and relative abundance levels of ARGs, respectively. The relative abundance values have been increased 106 times for better display.
The abundance levels of ARGs varied from bovine to bovine. Detailed information on the 42 ARG types and their relative abundance comparisons among yaks and dairy and beef cattle are outlined in Fig. 4: the percentage of one specific ARG in each group is equal to its corresponding abundance divided by the sum of the abundance of this ARG. Among these ARGs, bacA was the most abundant ARG and was nearly average in each individual. The bacA, also named uppP, which may be an intrinsic gene in bacteria, encodes an undecaprenyl pyrophosphate phosphatase and is responsible for bacitracin resistance. 36 Compared with bl2b_rob, bl1_ec and bl2e_cepA, the bl2e_cfxA gene was widely found in every individual and was the dominant beta-lactam gene in each individual (Supplementary Fig. S5a). However, although the abundance levels of those four genes differed greatly from bovine to bovine, bl1_ec and bl2e_cepA and bl2e_cfxA were major ARGs in dairy cattle, bl2b_rob was prevalent in beef cattle. The TER type tetQ had high abundance levels (top three) in each group, and among all the 10 tet types, tetQ possessed the highest percentage, followed by tetW (Supplementary Fig. S5b). Tet40 was the third most abundant TER in dairy and beef cattle, with nearly the same abundance as tetQ and tetW. However, in yaks, tet40 were 18 and 5 times less abundant than tetQ and tetW, respectively (Supplementary Table S3).
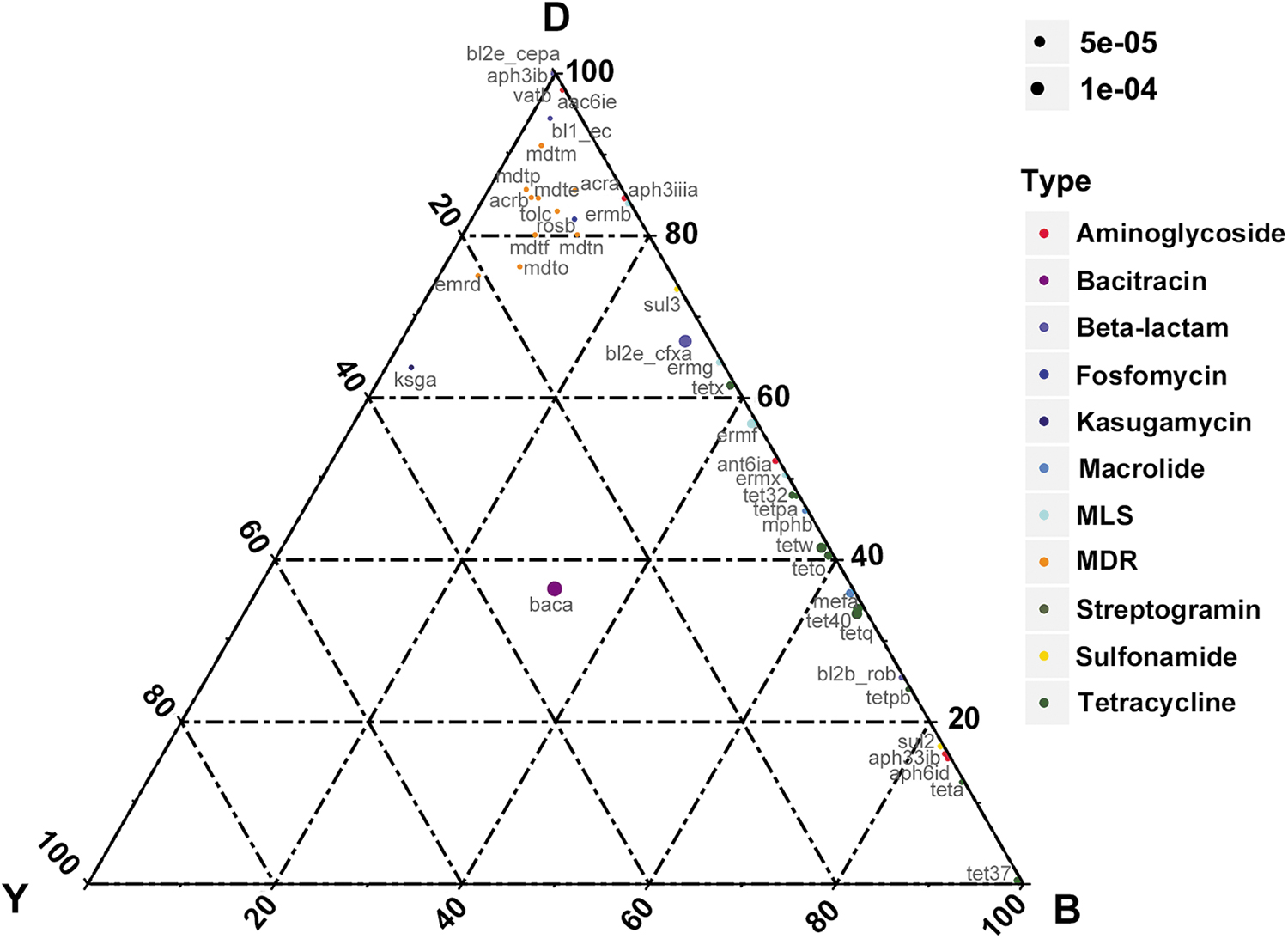
Ternary plot showing the relative abundance comparisons of 42 ARGs in the guts of yaks, dairy cows, and beef cattle. The sum of the abundance for one specific ARG in these three types of guts was set as 100%. In the ternary plot, the percentage (%) of one specific ARG in each gut is equal to its corresponding abundance divided by the abundance sum of this ARG in the three gut types. The symbol size indicates the relative ARG abundance. B = beef cattle; D = dairy cow; Y = yak.
Clustering of the abundance and single-nucleotide polymorphisms for ARGs
In this study, two different hierarchical clustering trees were structured to demonstrate the discrepancy of ARGs in the three groups. The heat map hierarchical clustering based on the relative abundance level of each ARG type showed that all the individual yaks could be separately clustered together, whereas the dairy and beef cattle could not (Fig. 5). An interesting result appeared in single nucleotide polymorphism (SNP) clustering. The SNPs on the ARGs identified in the yak were obviously different from those in the dairy and beef cattle farms, as was reflected in the neighbor-joining tree (Fig. 6). The SNP clustering could not divide the yak individuals between two distant regions, whereas most individuals from different farms could be clustered separately using this method.
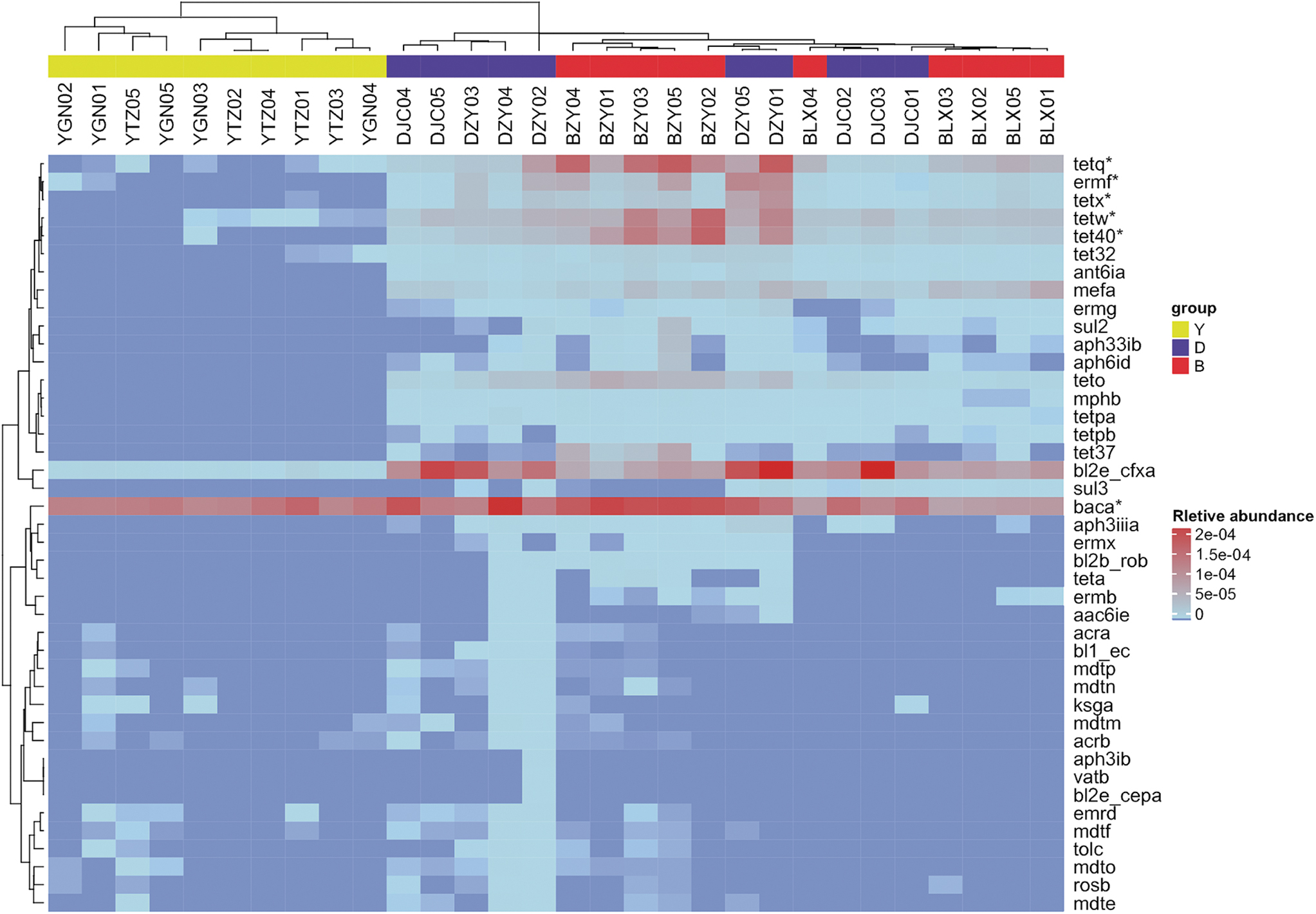
Heat map of the relative abundance level of each gene type in each individual and hierarchical cluster. The gene types (rows) and samples (columns) were clustered with the MultiExperiment Viewer (MeV version 4.6) using the Spearman rank correlation and complete linkage. *At least 60% of individuals harbored this gene type with a relative abundance of >1e-06.

Neighbor-joining tree structured using the SNPs in the ARGs from 30 individuals. Green = dairy cow; Red = beef cattle; Blue = yak. SNP, single nucleotide polymorphism.
Occurrence and abundance of MGEs
MGEs could transfer resistance determinants among microbes.37,38 In a BLAST search against our database, all the 472 IS types were found in our study, and the ISs showed diversity in different individuals. The relative abundance levels of IS elements were significantly higher in the dairy and beef cattle than in the yak (p < 0.0001) but were not considerably different between dairy and beef cattle groups (Fig. 7). This result was completely similar to that for the ARGs.

Comparison of the relative abundance levels of the MGEs (ISs) between yaks and dairy and beef cattle. The individuals are shown as dots on the left. The asterisks on the top indicate p < 0.0001 (Mann–Whitney test). The x-axis are sample group, and y-axis is the relative abundance of ARGs. B = beef cattle; D = dairy cow; Y = yak. MGE, mobile genetic element.
Discussion
The large-scale breeding of food animals has become a trend in modern agricultural production, with the goal of providing sustainable nutrition supplies, such as eggs, milk, and meat. Closed breeding is an effective method, but because of the high animal density, infectious diseases are easy to disseminate among animals.39,40 A large variety of antimicrobials have been widely used to fight bacterial infectious diseases.17,41 Misuse and abuse of antimicrobials are bound to result in antibiotic residues and the selection of resistant bacteria. Consequently, it is almost impossible to find a pristine modern environment that has never been contaminated with anthropogenic antimicrobials. 42 In contrast, yaks live on unpolluted highland pastures 43 that are devoid of anthropogenic impacts, and antimicrobials are hardly used. Thus, the yak is a natural control to study the effects of antimicrobials on ARGs in the animal gut microbiota.
This study provided a comprehensive overview of the ARG reservoir in the bovine gut microbiota at the metagenomic level and analyzed the effects of the antimicrobials used for ARG reservoirs. The diversity and abundance levels of ARGs detected in closed breeding farms (dairy and beef cattle) were significantly higher than those detected in yaks, indicating that ARGs were commonly prevalent under these selection pressures. Antimicrobial consumption is a major reason for the emergence of resistance, and the differential selection pressures of antimicrobials lead to differences in drug resistance. 44 Undoubtedly, antibiotics are enormously important for humans and efficient food animal production and can be effective at preventing and treating pathogenic infections. 45 However, antimicrobial overuse and abuse are severe in China. 16 Furthermore, some classes of antibiotics used in humans have also been used in animals. 46 These practices are considerable and serious, as antimicrobial use in food animals could select for resistant bacteria, including pathogens. As a result, when these pathogens infect humans, they will increase the burden of illness, resulting in a huge impact on health. 47
Furthermore, the relationship between ARG emergence and the usage of antimicrobials is very complex. 16 As we have learned from the herdsman, only a small amount of penicillin is used against disease in individual yaks. However, our results showed that despite their low levels of abundance, a diverse collection ARGs was found in yaks, such as TER, beta-lactams genes. Previous studies have reported that bacteria can obtain resistance to some older antimicrobials (e.g., penicillin, tetracycline, streptomycin, and sulfonamide) without any contact with these drugs. 48 According to Jakobsson et al., 49 resistant bacteria and ARGs can persist for years, even under short-term antibiotic administration. These reasons may explain why TER elements were diverse.
In addition, it is noteworthy that ARGs can be horizontally transferred between microbes through MGEs. 50 The common prevalence of tetracycline and beta-lactam resistance genes is most likely owing to HGT by MGEs.14,50 The HGT of ARGs can occur between nonpathogens, pathogens, and even distantly related organisms. Recent studies have demonstrated specific instances of the HGT of ARGs in human and animal guts, soil, sediments, and water.2,3,17,51,52 Moreover, the HGT of ARGs is more common than that for other genes and is 25 times more likely to be found in the gut than in other environments. 53 In this study, ARGs were found in high abundance in the environment, with correspondingly higher abundance levels of MGEs (ISs), which was a factor in the prevalence of ARGs in closed breeding farms.
Nevertheless, it should be noted that the long-term use of antibiotics is the strongest determinant for ARG prevalence. The discovery and use of penicillin in the past 60 years have forewarned of the resistance that bacteria could develop to antimicrobials over time. 45 Penicillin is still one of the most popular antibiotics in veterinary clinics. Thus, it is clear why the bl2e_cfxA gene is widespread. Moreover, hundreds of ARG subtypes that would diminish the effectiveness of beta-lactam antibiotics have been detected. 54 However, the diversity and high abundance of ARGs in closed breeding farms warn that we could soon be powerless to prevent disease when various ARGs are prevalent in pathogens.
In conclusion, this study is the first comprehensive demonstration of the ARG reservoirs at different bovine farms. The results suggest that the bovines in closed breeding farms harbor richer reservoirs for the accumulation and dissemination of ARGs than yaks and caution against the group-level use of antimicrobials. The reasons for this phenomenon may be complex and diverse. Of note, diversity and excessive use of antimicrobials in each of the closed breeding farms should play major roles in altering the resistant microbiota and ARG transmission. Additional large-scale studies, PacBio or Nanopore technology, would give a lot more details of the MGEs and plasmids to analyze the prevalence and fate of ARGs.
Accession Numbers
All 30 gut metagenome sequences have been deposited in the NCBI SRA database under the accession numbers SRX2972971 to SRX2973000.
Ethics Approval and Consent to Participate
Permission for accessing specific locations, information regarding the number of samples harvested, and an associated permit number for calves were not required, and no endangered or protected species were involved or harmed during this study.
Consent for Publication
All the authors agreed to the publication of the article.
Availability of Data and Material
The data supporting the findings of this study are contained within the article.
Footnotes
Authors' Contributions
Z.Z. and J.Y.Z. designed the study; Z.Z., M.Z.C., Y.X.S., Y.Z., T.H.M., G.H. L., X.C., and Z.X.Z. generated and provided the dataset; Z.Z., L.W.Z., and W.W.W. performed the experiments, analyzed the data, and wrote the article.
Acknowledgment
The authors thank the State Key Laboratory for Infectious Disease Prevention and Control (China CDC) for providing funding and technical support.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (No: 31872520); Drug Development and Clinical Drug Use Posts of National Beef Yak Industry Technical System (No: CARS-37); Hebei Natural Science Foundation (C2019402114).
Disclosure Statement
The authors declare that they have no competing interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.