
Abstract
Recent genomic studies of methicillin-resistant Staphylococcus aureus (MRSA) have revealed genetic diversity in the various clonal lineages. Along with clinical concerns of MRSA infection, infection with heterogeneous vancomycin-intermediate S. aureus (hVISA) is closely associated with treatment failure. In this study, we investigated the magnitude of genetic variation and features at the genomic level of hVISA strains isolated in South Korea. Four hVISA strains were analyzed by molecular epidemiology, antimicrobial susceptibility, and whole-genome sequencing methods, and they were compared with the reference VISA and vancomycin-susceptible S. aureus strains in the same clonal lineage. The epidemiologic features of hVISA strains were closely related to the ST5 and ST239 clones. Comparative analysis of the whole genome showed genetic mutations, particularly in two-component systems (TCSs) and transcriptional regulators. Genetic mutations in walK were commonly found in both ST5- (F545L, E378K, T500K) and ST239-related (E424D, T492R) hVISA strains. hVISA strains in the ST5 clonal lineage contained mutations in TCS genes, including the walK, vraR, and agr loci, whereas ST239-related strains harbored different genetic variations in walK, lytR, and saeR. This study suggests that the diverse genetic variation of TCSs and transcriptional regulators are involved in reduced vancomycin susceptibility through different mechanisms in each clonal lineage.
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) is an important bacterium causing hospital- and community-associated infections ranging from benign skin and soft tissue infections to fatal invasive diseases.1–3 Increasing antibiotic resistance is a severe threat to humans. Particularly, the emergence of vancomycin-intermediate S. aureus (VISA) and heterogeneous-VISA (hVISA) frequently results in treatment failure and persistent infection.4–6 VISA and hVISA strains are often isolated from patients with an underlying disease such as malignancy or after a prolonged period of infection.7–10
Vancomycin heteroresistance can be caused by a mechanism that alters cell wall turnover, autolytic activity, and cell wall synthesis, thereby leading to cell wall thickening and reduced vancomycin susceptibility.11–13 Acquisition of vancomycin heteroresistance likely occurs through diverse and complex pathways, even among closely related strains. 7 Regarding changes in cell wall synthesis in VISA/hVISA strains, diverse molecular mechanisms likely participate in reducing vancomycin susceptibility rather than introducing a vancomycin resistance gene. These molecular mechanisms mainly involve transcriptional changes such as upregulation of cell wall stimulon genes14,15 and mutations in genes associated with cell wall synthesis regulation.16–18 Indeed, diverse mutations in global regulators of S. aureus rather than a specific mutation have been implicated in the emergence of reduced susceptibility to vancomycin.17–19
Molecular epidemiological studies have shown that many hVISA and VISA strains are closely related to clonal complex (CC) 5 (mainly ST5) or CC8 (mainly ST239), of which strains are successfully adapted in the hospital.19,20 Epidemiological studies in South Korea have shown that the ST5 and ST239 strains are predominant in hospital-associated infections, and hospital-associated hVISA/VISA strains belonged mainly to these clonal lineages.21–25 However, these studies were limited to the molecular epidemiological features of hVISA/VISA strains, whereas their genetic features remain unclear.23–25
In this study, we analyzed the genetic mutations at the genomic level by whole-genome sequencing (WGS) in hVISA strains of South Korea. We also analyzed the differences in genes related to antimicrobial resistance in the different clonal lineages and accumulated genetic mutations associated with vancomycin heteroresistance. In addition, we compared the genomic profiles of hVISA with those of vancomycin-susceptible S. aureus (VSSA) and reference VISA strains.
Materials and Methods
Bacterial strains
Four hVISA strains (V521, V605, NCCP 14562, and NCCP 14558) were obtained from the Korea Disease Control and Prevention Agency (Osong, South Korea) and National Culture Collection for Pathogens (NCCP). These strains were isolated from specimens of wound infection and catheter-borne infection. Mu3 (ATCC 700698, Manassas, VA) hVISA, Mu50 (ATCC 700699) VISA, and S. aureus ATCC 29213 VSSA strain were used as controls for performing antimicrobial susceptibility analysis and population analysis profiling (PAP). Each strain was grown on tryptic soy agar/broth (TSA; BD Biosciences, Frankiln Lakes, NJ) and Müller-hinton broth (BD Biosciences) for antimicrobial susceptibility testing.
Genomic DNA
The genomic DNA was purified from these strains by using a Masterpure Gram Positive DNA Purification Kit (Epicentre, Madison, WI) according to the manufacturer's instructions, except that 100 U/mL of lysostaphin (Sigma-Aldrich, St. Louis, MO) was added during the lysis step. After DNA extraction, DNA concentration and purity were investigated with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Seoul, Korea). When A260/280 and A260/230 ratio were more than 1.80 and 2.00 to 2.20, respectively, genomic DNA extracts were verified as being of high purity.
Antimicrobial susceptibility
The antimicrobial susceptibilities of the VISA strains were determined by using the broth dilution method, as recommended by the Clinical and Laboratory Standards Institute (2010). 26 The minimum inhibition concentration (MIC) of daptomycin (DAP, Sigma-Aldrich), linezolid (LZ; Sigma-Aldrich), trimethoprim-sulfamethoxazole (TMP/SMX; Sigma-Aldrich), and vancomycin (VAN; Sigma-Aldrich) was measured by the broth microdilution assay. Vancomycin PAP was performed as previously described. 27 The experiments were repeated at least four times for each strain. The area under the curve (AUC) ratio of each strain was calculated by comparison with the Mu3 (ATCC 700698) AUC. When the AUC ratios were ≥0.9 and ≥1.3, the strains were identified as hVISA and VISA, respectively. 27
Molecular epidemiology
Pulse field gel electrophoresis (PFGE) was performed as previously described. 28 S. aureus NCTC 8325 strain was used as a reference standard in the PFGE analysis. Using a previously established database, 22 the clonality of VISA or hVISA strains was determined based on a similarity cutoff of 80% (analyzed by the dice coefficient and unweighted pair group method with arithmetic mean, with 1% tolerance and 0.5% optimization settings). Multilocus sequence typing (MLST) was performed as previously described, 29 and each sequence type (ST) was assigned based on the S. aureus MLST database. We performed NCBI genomic BLAST (Microbial Genomes BLAST) for all complete genomes of “Staphylococcus aureus (taxid:1280).” We downloaded the genomic BLAST results of comparison among genomes of closely related VSSA and VISA strains with that of S. aureus NCTC 8325 as an outgroup (S. aureus NCTC 8325, Assembly no. GCA_000013425; N315, GCA_000009645; Mu50, GCA_000009665; Mu3, GCA_000010445; JH1 GCA_000017125; T0131, GCA_000204665; JKD6008, GCA_000145595; Z172, GCA_000485885; TW20, GCA_000027045).
WGS and analysis
Whole-genome sequences were generated for each strain by using PacBio RS II with P5–C3 chemistry (Pacific Biosciences, Menlo Park, CA). The minimum polymerase read quality was 0.8, and the mean subread lengths of each single-molecule real-time cell were more than 1 Kb. PacBio-only de novo assembly was performed by using SMRT Analysis v2.3.0 HGAP.2 protocol 30 (Supplementary Table S1). HGAP implements a hierarchical assembly process for a single long pass read generated by the PacBio sequencer, which involves three main steps: pre-assembly, assembly, and consensus polishing. The pre-assembly step generates long and highly accurate sequences by mapping single reads to seed reads longer than the rest and subsequent error correction by quality trimming. The assembly step uses the overlap-layout-consensus approach for pre-assembled reads. In the last step, the consensus-calling algorithm Quiver enhances accuracy significantly by using local realignment and quality scores. To evaluate both ends of the circular contigs and trim the overlapping regions, we used gepard software package and minimus2 in the AMOS package. The orientation and origin of circular sequences were adjusted according to the genome sequence of the reference strain N315 (GenBank no. BA000018).
We have submitted these sequences to Sequence Read Archive and GenBank. All sequencing data used in this work are available from these databases (CP013953; CP013955; CP013957; CP013959). We annotated the sequences by using the NCBI Prokaryotic Genome Annotation Pipeline. After annotation, we created protein blast DB of all protein-coding open reading frames of 8 strains and performed local blast to identify corresponding proteins in each strain and in the reference strain.
The genomes of hVISA strains in this study were compared with those of the reference VSSA and VISA strains (S. aureus N315, GenBank no. BA000018; Mu50, BA000017; TW20, FN433596; Z172, CP006838). We performed multi-genome alignment with the progressiveMauve algorithm 31 by using the DNASTAR megalign pro software (Dnastar, Inc., Madison, WI). This method investigated the recombination, including rearrangement, segmental duplication, gain, and loss in the same clonal lineage (ST5 clonal lineage; NCCP 14562 & 14558 vs. VSSA N315 & VISA Mu50, ST239 clonal lineage; V521 & V605 vs. VSSA TW20 & VISA Z172). It defined a pairwise locally collinear block as a subset of local alignments in a pair of genomes. It built up genome alignments according to a distance matrix and a guide tree computed based on an estimate of each pair of shared gene content. At each step of the genome alignments, alignment anchors were selected, and an anchored profile–profile global alignment was performed with a modified MUSCLE method.31,32 A parameter of seed weight was 15.
Insertion sequences and prophage regions of genomes were identified by using PHAST 33 and ISfinder. 34 We compared the integrase of each phage with those of phage genomes from the NCBI nucleotide database and analyzed them by the integrase groups, as previously described.35,36 Antibiotic resistance-related genes were explored in each genome by using the Comprehensive Antibiotic Resistance Database. 37 We investigated the protein–protein interaction at the STRING database. 38 Also, the pathway of each gene was obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG). 39
Results
Antibiotic susceptibility
The VAN MIC of all hVISA strains in this study was 4 mg/L, and PAP revealed that all hVISA strains are heterogeneous VAN intermediate susceptible, as the AUC ratio of NCCP 14558, NCCP 14562, V521, and V605 was 1.13, 1.07, 1.06, and 1.20, respectively. The range of DAP and LZ MIC in these strains was 4–8 mg/L and 0.5–2 mg/L, respectively. The TMP/SMX MIC of the NCCP 14558 and 14562 strains was 2 and 0.25 mg/L, respectively. However, the MIC of V521 and V605 was 64 mg/L.
Epidemiological features
Analysis of the PFGE patterns showed that strains NCCP 14558 and NCCP 14562 were clustered with ST5 clinical VSSA isolates. Stains V521 and V605 were grouped into the ST239 cluster (similarity >80%). The hVISA strains in this study were predicted to be closely related to two clonal lineages (ST5, ST239), and the clinical isolates in South Korea were predominant in hospital-associated infections (Supplementary Fig. S1). In addition, MLST showed that both strains NCCP 14558 and NCCP 14562 were ST5 (allelic profile, 1-4-1-4-12-1-10). V521 and V605 were ST239 (2-3-1-1-4-4-3) and ST5355 (ST239 single locus variant, 2-3-1-238-4-4-3), respectively.
Genomic features of hVISA strains
The length of the largest contigs was more than 2.9 Mb, which was comparable to the genome size 3 Mb of the known reference N315. WGS of strains NCCP 14558, NCCP 14562, V521, and V605 showed sizes of 2.95, 2.911, 3.086, and 3.089 Mb, respectively. The result of genomic BLAST exhibited that NCCP 14558 and NCCP 14562 strains are closely related to reference VISA and VSSA strains (N315 and Mu50), and V521 and V605 are clustered into the group of TW20 and Z172 strains (Fig. 1). The general genomic features of these hVISA strains are summarized and compared with those of the reference strain in Table 1.
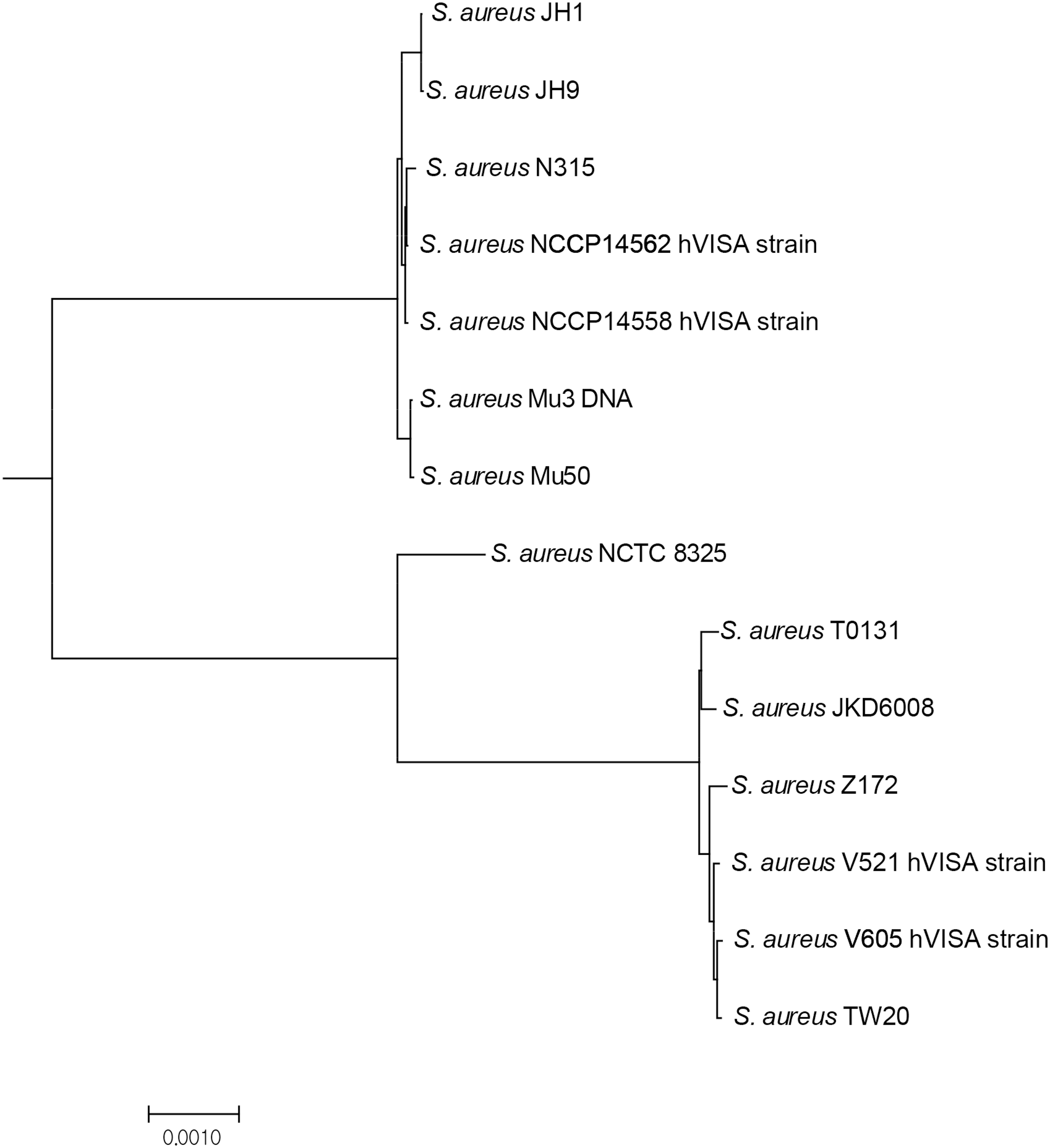
A phylogenetic analysis in genome sequences of closely related reference stains (S. aureus NCTC 8325, Assembly no. GCA_000013425; N315, GCA_000009645; Mu50, GCA_000009665; Mu3, GCA_000010445; JH1 GCA_000017125; T0131, GCA_000204665; JKD6008, GCA_000145595; Z172, GCA_000485885; TW20, GCA_000027045) with those of hVISA strains in this study by NCBI genomic BLAST (Microbial Genomes BLAST) for all complete genomes of “Staphylococcus aureus (taxid:1280)”. A phylogenetic tree was drawn by mega version 6.0 software using a genetic distance data obtained from genomic BLAST. hVISA, heterogeneous vancomycin-intermediate Staphylococcus aureus.
Comparison of General Features in Heterogeneous Vancomycin-Intermediate Staphylococcus aureus and Reference Strains
Numbers in parentheses indicate GC %.
SCC, staphylococcal cassette chromosome; hVISA, heterogeneous vancomycin-intermediate Staphylococcus aureus.
Using BLAST and IS finder, the genomic islands in each strain were analyzed by comparison of the whole genomes in the same clonal lineage (ST5; NCCP 14562, NCCP 14568, N315, Mu50, ST239; V521, V605, Z172, TW20). Except for Tn5801, there were no important differences in the genomic islands of the ST5 lineage strains. The hVISA and Mu50 strains harbor Tn5801, including tetM, conferring resistance against tetracycline. No differences were observed among the ST239 lineage strains. Taken together, these findings indicated that there were a few differences between the VSSA and VISA strains in the gene profile associated with genomic islands (Table 1). Analysis of the staphylococcal cassette chromosome (SCC) region revealed that ST5 lineage strains harbored SCCmec II, whereas ST239 lineage strains harbored SCCmec III and SCCmercury (Table 1). Comparative analysis of the overall genomic structure by the mauve method showed that each strain had a different prophage location, number, and type (Table 1 and Fig. 2A, B). Their respective prophage regions in the genome were mostly unique to each strain, despite that these strains were from the same clonal lineage. The integrase analysis exhibited unique characteristics of the prophage profile in each strain (Supplementary Fig. S2). In the ST239 clonal lineage, three prophage regions (ΦSa1, ΦSa3, and ΦSaβ) were homologous in each strain, whereas ΦV521 (Sa 5 integrase group, located at 2034171–2097539) and ΦV605 (Sa 7 integrase group, 1195898–1256003) of hVISA were unique (Fig. 2B and Supplementary Fig. S2). In the ST5 clonal lineage, there was a significant difference in the prophage profile (Supplementary Fig. S2). Prophages on the genome of hVISA strain (NCCP 14558 & 14562) belong to Sa integrase 2 (Sa 2), Sa 5, and Sa 7 group, whereas ΦMu50A and ΦMu50B in Mu50 VISA strain belong to Sa 3 and Sa 1 group, respectively.
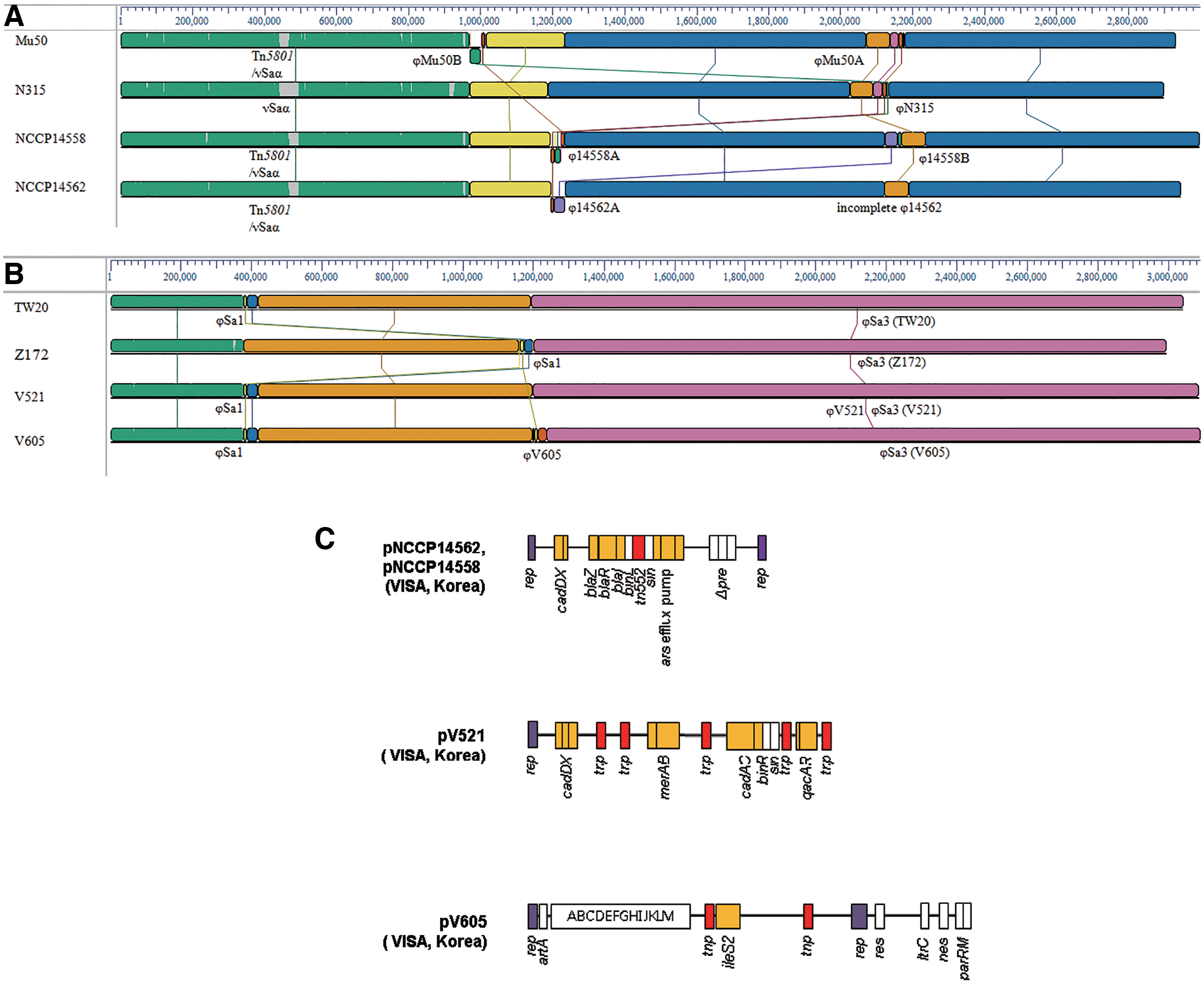
Comparative genome analysis and major structural features of hVISA plasmids. Comparative genome analysis was performed in the genomes of hVISA, the reference VISA (NCCP 14562, NCCP 14558, V521, V605, Mu50, & Z172), and VSSA (N315 and TW20) strains with ST5
Comprehensive analysis of antibiotic resistance-related genes (including efflux pump, hydrolyzing enzyme, and antibiotics target variant) at the genome level revealed no difference in each hVISA strain of the same clonal lineage. In contrast, a different profile of antibiotic resistance-related genes was observed between the ST5 and ST239 lineage strains (Supplementary Table S2). The ST239 lineage hVISA strains (V521 and V605) contain three classes of aminoglycoside resistance genes (AAC(6′)-Ie-APH(2′′)-Ia, ANT(9)-Ia, APH(3′)-IIIa), whereas ST5 hVISA strains harbor only one class of aminoglycoside resistance gene (ANT(9)-Ia) (Supplementary Table S2). In addition, the profile of the TMP resistance gene showed that ST239 strains resistant to TMP harbored dfrC and dfrG, whereas ST5 strains susceptible to TMP contained only dfrC (Supplementary Table S2).
Antibiotic resistance genes and mobile elements in plasmids
The NCCP 14562 and NCCP 14558 hVISA strains harbored the plasmids pNCCP14562 (24,657 bp, GenBank accession no.CP013956.1) and pNCCP14558 (24,729 bp, GenBank accession no.CP013954.1), respectively. Both plasmids contained the penicillin-resistance module “blaZ-blaR1-blaI-binL” bracketed with Tn522 insertion sequences, which were highly similar to those in the pN315 (AP0031339.1) and CA-347 (CP006045.1) plasmids from MRSA isolates.40–42 Both plasmids contained the cadmium resistance gene cadDX and arsenate resistance gene arsRBC (Fig. 2C, Supplementary Table S3).
V521 hVISA strain harbored pV521 (28,723 bp, GenBank accession no.CP013958.1), which was highly similar to the plasmid pZ172_1 (CP006839.1) in a VISA isolate from Taiwan. 43 The cadmium resistance genes cadDX and merAB confer high levels of tolerance to organomercurial compounds, and multi-drug efflux pump qacAR genes are involved in resistance to metals and antibiotics of pV521 (Fig. 2C and Supplementary Table S3). The plasmid pV605 (43,115 bp, GenBank accession no.CP013960.1) from V605 showed highly similar sequences to pV030–8 (CP006839.1) and pPR9 (CP006839.1) from MRSA strains in South Korea and Spain, respectively (Fig. 2C and Supplementary Table S3). 44 pV605 carried ileS2 flanked by IS257, contributing to high-level mupirocin resistance in staphylococci. However, the highly conserved fragment “blaZ-blaR1-blaI” found downstream of IS257 in pPR9 was lacking in pV605. The insertion sequence IS257 may play an important role in rearrangement of pV605. The plasmids in hVISA isolates from South Korea carried various resistance genes to antimicrobials and metals, contributing to antibiotics and metallic resistance.
Mutation profile of two-component systems, gene regulator, antibiotics resistance, and cell wall proteins between the same clonal lineage
We detected polymorphisms in two-component systems (TCS), gene regulators, antibiotics resistance, and cell wall proteins genes related to reduced vancomycin susceptibility compared with those in the reference VSSA strains (N315, TW20) and VISA strains (Mu50, Z172) in the same clonal lineage. In the ST5 VISA strains (NCCP 14558, NCCP 14562), four mutations were found in TCS genes, including walK, vraSR, agr, and yesM (Table 2). In addition, mutations were found in four regulator genes, including transcriptional regulation such as lysR and sarU (Table 2). Mutations were detected in antibiotic resistance-related genes and cell wall proteins, including DNA-directed RNA polymerase beta chain (rpoB), penicillin-binding protein 2 (pbp2), fluroquinolone target gene (gyrA), and LPXTG-motif cell wall protein (isdB) (Table 2).
Mutation Profile of Two-Component Systems, Transcriptional Regulators, Cell Wall Metabolism, Autolysis, and Antimicrobial Resistance-Related Gene in ST5 Vancomycin-Intermediate Staphylococcus aureus Strains (NCCP 14558 & NCCP 14562) Compared with Those of N315 Vancomycin-Susceptible Staphylococcus aureus Strain
It indicates orf number of S. aureus N315 strain (NC_002745).
TCS, two-component system.
In ST239-related hVISA strains (V521 and V605), genetic variations were found in five TCS genes: walK, lytR, saeS, yhcR, and kdpD (Table 3). Mutations were also found in two genes of another regulator, including transcriptional regulators such as merR, araC, and recX (Table 3). Mutations were detected in the antibiotic resistance-related genes fmhA, penicillin-binding protein 1 (pbpA), oxacillin resistance-related membrane protein (fmtC), and LPXTG-motif cell wall protein (isdA, isdB) (Table 3).
Mutation Profile of Two-Component System, Transcriptional Regulators, Cell Wall Metabolism, Autolysis, and Antimicrobial Resistance-Related Gene in V521 and V605 Vancomycin-Intermediate Staphylococcus aureus Strains Compared with Those of TW20 Vancomycin-Susceptible Staphylococcus aureus Strain
It indicates orf number of S. aureus TW20 strain (NC_017331).
Mutation profiling of transcription regulators showed that mutations in walK were common in the analyzed VISA strains of both clonal lineages (ST5 and ST239), whereas diverse differences in walK were observed in each strain. The F545L mutation was found in walK of NCCP 14558. E378K and T500K were found in walK in NCCP 14562. E424D and T492R were found in walK of V521 and V605, respectively. Diverse mutations were detected in a global regulator, including a cell wall synthesis regulon and transcriptional regulators (Tables 2 and 3). The protein–protein network analysis showed that vraSR, walKR, and graSR are interactively involved in cell wall metabolism, and diverse mutations in this network could cause the change of cell wall synthesis and autolysis, resulting in the vancomycin-intermediate resistance (Fig. 3A).
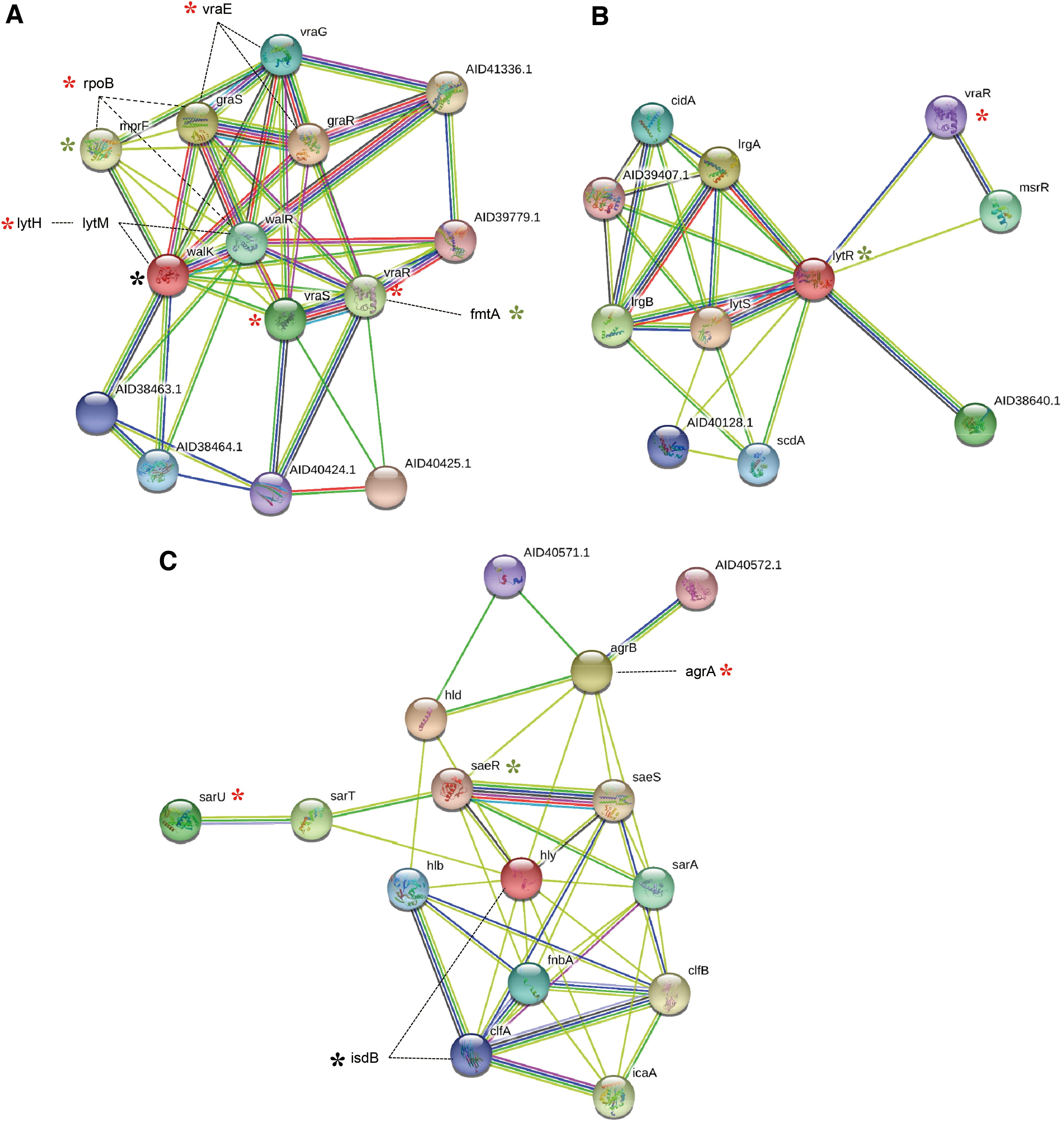
The plot of protein network in TCS, transcriptional regulators, cell wall metabolism, autolysis, and antimicrobial resistance-related genes of Staphylococcus aureus. The basic plot of the protein–protein interaction was obtained from the data of STRING server. We additionally illustrated some genes with the mutational change in the antimicrobial resistance, cell wall metabolism, and autolysis-related network.
Discussion
Reduced vancomycin susceptibility often results in therapeutic failure and persistent infection. We investigated the diverse genetic changes in hVISA strains in South Korea at the genomic level for comparative analysis among reference VSSA and VISA strains from the same clonal lineage.
Molecular epidemiology evaluation (MLST and PFGE) showed that the hVISA strains evaluated in this study were closely related to clinical MRSA isolates (VSSA) in two clonal lineages of ST5 and ST239 (Supplementary Fig. S1). For comparative studies, genomic BLAST analysis demonstrated that NCCP 14562 and NCCP 14558 were similar to international ST5 lineage strains, such as N315 VSSA and Mu50 VISA strains, and V521 and V605 was similar to international ST239 lineage strains, such as TW20 VSSA, multi-antibiotic- and antiseptic-resistant MRSA strains 36 and Z172 VISA strains (Fig. 1). It is assumed that these hVISA strains evolved from the predominant MRSA strains of a clinical setting in South Korea and were closely related to clonal lineage VISA strains dispersed worldwide.
Comparative analysis of the overall genomic structure of these hVISA strains showed that the class and distribution of their genomic islands were very similar to those in the reference VISA and VSSA strains of the same clonal lineage, except for the presence of Tn5881 (tetM) in VISA and hVISA strains (Mu50, NCCP 14562, and NCCP 14558) (Table 1 and Fig. 2). In addition, the distribution of antibiotic resistance-related genes, such as those encoding antibiotic hydrolyzing enzymes and efflux pumps, and mutations in the targets of antibiotics, were very conserved in the same clonal lineage; in contrast, there were a few differences, such as the class of aminoglycoside resistance-related genes and TMP resistance-related genes (dfrG-harboring strains are TMP-resistant) 45 between the ST5 and ST239 lineage strains (Supplementary Table S2). Challagundla et al. reported a phylogenomic result of CC5 MRSA strains in the Western Hemisphere, and there were three clades (CC5-Basal, CC5-I, and CC5-II) of MRSA. 46 CC5-I MRSA had more antibiotic resistance and fewer toxins, and CC5-II MRSA had more antibiotic resistance and virulence than other MRSA clones. 46 These were distinct from the ST5-II N315 (CC5-Basal clade) strain from Asia (Japan). 46 In this study, NCCP 14558 and NCCP 14562 (ST5, inserted with Tn916 subfamily Tn5801) hVISA strain had a similar profile for antibiotics resistance, genomic islands, SCC mec type, and transposons with Japan ST5-II VSSA (N315, not inserted with Tn5801) and VISA (Mu50, Tn5801) strain, except for tetracycline and vancomycin resistance (Table 1). In terms of transposons and antibiotic resistance, ST5 hVISA strain in this study should be much closer to ST5 Japan strain (N315 & Mu50) than CC5-I and CC5-II MRSA strains in the Western Hemisphere. However, there was a significant difference in the prophages profile between hVISA (NCCP 14558 & NCCP 14562) and Japan ST5-II (N315 & Mu50) strains. There were prophages of other Sa integrase groups (Sa2, Sa5, and Sa7 groups) in hVISA strain, instead of ΦSa 3 family harbored in N315 and Mu50 (Table 1 and Supplementary Fig. S2). These findings suggest that hVISA strains should be sub-diverged from ST5 Japan strains, not from CC5-I or CC5-II in Western Hemisphere.
Also, plasmids of these hVISA strains in this study showed high similarity with those of VISA and MRSA strains worldwide. The plasmids pNCCP14562 and pNCCP14558 in the NCCP 14562 and NCCP 14558 isolates, respectively, were very similar to pN315 in strain N315. pV521 and pV605 (plasmids of V521 and V605, respectively) also shared highly similar sequences with a plasmid in the Z172 VISA strain from Taiwan. Analysis of plasmids in the hVISA strains was consistent with those of their host strains, suggesting that VISA or MRSA found worldwide were rather introduced into South Korea via the transmission of plasmids carrying resistance to antibiotics and metal into S. aureus.
Unlike the similarity observed in the genomic structure, the plasmid, and antibiotic resistance-related genes, the comparative profiling study of genetic changes showed that TCSs, transcriptional regulators, and cell wall synthesis-related genes have unique characteristics (Tables 2 and 3). Specifically, TCSs, consisting of a sensor histidine kinase and cognate response regulator, can affect the susceptibility to several antibacterial agents by regulating the expression of genes related to cell wall/membrane synthesis, autolysis, and others. Regarding vancomycin susceptibility, diverse genetic variations in walKR have been reported in most VISA or hVISA strains worldwide,47,48 which are predicted to be involved in the reduction of vancomycin susceptibility. In this study, mutations in walK were found in both ST5- and ST239-related hVISA strains (Tables 2 and 3). However, in addition to walK, genetic variation in other TCSs and cell wall/membrane synthesis- and autolysis regulation-related genes differed between clonal lineages (ST5, ST239) (Tables 2 and 3). In ST5 hVISA strains (NCCP 14558 and NCCP 14562), we noted the mutations of vraSR and rpoB with that of walK. These mutations of vraSR, rpoB, and walK are frequently associated with hVISA/VISA development.47,49,50 However, in ST239-related hVISA strains, there were no mutations of vraSR, and the mutation of lytR interacted with vraR (Fig. 3B). LytSR regulates genes involved in autolysis and cell wall metabolism through regulation of lrgA and lrgB. Also, lytR is associated with msrR (Fig. 3B), belonging to the LytR-CpsA-Psr (LCP) family involved in cell wall metabolism, autolysis, and antibiotics susceptibility. 51 Agr and SaeSR are global regulators that can upregulate and downregulate adhesion molecules, toxins, and other virulence factors with networking. In the Agr-SaeSR network, there are a few mutational changes in all hVISA strains (Fig. 3C). Agr dysfunction was reported to be potentially associated with hVISA or VISA.23,52
These findings suggest that the accumulated mutations in the diverse cell wall metabolism associated regulators result in reduced vancomycin susceptibility, and these changes may be derived differently in their own way, depending on clonal lineages. In this study, there were some limitations that we did not investigate, for example, the genetic effects of the mutations on the vancomycin susceptibility and protein–protein interaction. However, the information on the diverse mutations could provide benefits to understand the molecular nature of heterogenous antimicrobial resistance. Also, profiling of the diverse genetic variation involved in cell wall metabolism-associated network may be valid for the proper diagnosis, surveillance, treatment, and prevention of hVISA in the genome era.
Footnotes
Acknowledgments
The authors acknowledge the support of the Korea Centers for Disease Control and Prevention (Osong, South Korea) and NCCP (Osong, South Korea) for providing hVISA strains. This study was approved by the institutional review board at Seoul St. Mary's Hospital (approval no. KC15SISI0510).
Disclosure Statement
All authors (C.P., D.G.L., K.R., J.Y.S., S.Y.C., and Y.J.C.) declare that they have no conflict of interest in connection with the submitted manuscripts.
Funding Information
This work was supported in part by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (No. HI14C2658).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.