
Abstract
Azole resistance constitutes a serious clinical problem in the management of infections caused by Candida albicans. This study aimed to explore azole-resistant mechanisms in clinical C. albicans isolates collected in Jinan, Shandong, China. In total, 22 samples were collected and analyzed. Among these, four isolates (28A, 28D, 28I, and 28J) exhibited high level of pan-azole-resistance that was Hsp90 dependent. Gene sequencing revealed that the four Hsp90-dependent strains contained different ERG3 mutations that led to four novel amino acid substitutions (S265Y, N322D, N324S, and E355D) in Erg3. The role of these substitutions in azole resistance development was determined by constructing one copy of the mutated ERG3 from the 28A, 28D, and 28I strains into C. albicans CAI4, respectively. The minimum inhibitory concentration value of fluconazole (FLC) against C. albicans CAI4-ERG328I increased fourfold compared with the wild-type C. albicans strain, suggesting that the novel combination of substitutions S265Y, N322D, and N324S played an important role in mediating azole resistance in 28I. Besides, we identified several different mechanisms in other three isolates. Strains 28A and 28D displayed increased efflux ability and overexpression of MDR1. Strain 28J showed high level of ERG11 expression, but no mutation in its regulator Upc2 was observed. Our study revealed that multiple factors confer azole resistance in clinical C. albicans isolates and combination therapy should be conducted clinically.
Introduction
C
To date, several mechanisms have been considered to underpin the azole resistance in C. albicans. The most common azole resistance mechanism is the upregulation of efflux transporters, including the ATP binding cassette (ABC) family members Cdr1 and Cdr2, and the major facilitator superfamily (MFS) proteins, such as Mdr1. 5 Another prevalent resistance mechanism is mutations in the ERG11 gene, which lead to amino acid substitutions in lanosterol 14α-demethylase (Erg11), decreasing the binding affinity of azole drugs to their target enzyme.6,7 Overexpression of ERG11 can also result in azole resistance. 8 In addition, mutated ERG3 encodes inactive sterol △5,6-desaturase and contributes to azole resistance by causing alterations in the ergosterol biosynthetic pathway. 9 In fact, multiple mechanisms usually combine to contribute to azole resistance in a single isolate.
In this study, we determined the susceptibility profiles of 22 clinical C. albicans isolates collected from 7 cancer patients to 5 different azole drugs and characterized 4 isolates (28A, 28D, 28I, and 28J) that exhibited resistance to a wide range of azoles. The alterations in the expression levels of azole-targeted enzyme Erg11, mutations in genes ERG3 and ERG11 as well as their regulators UPC2, the activity of the efflux pumps, and the mutations in their regulators CAP1 and MRR1 in these strains were examined to identify new mechanisms.
Materials and methods
Strains and growth conditions
In total, 22 isolates of C. albicans recovered from the oral buccal mucosal surfaces of 7 cancer patients at 2 affiliated teaching hospitals of Shandong University in Jinan (China) from 2008 to 2009 were analyzed in this study. All the patients have been previously diagnosed of advanced cancer for more than 1 year and accepted chemotherapy treatment.10,11 The strains were identified as Candida by CHROMagar based on color change and colony morphology and further confirmed by API20C (BioMerieux France) based on carbon source utilization and other differential biochemical assays according to the manufacturer's instructions. These strains were cultured on yeast–peptone–dextrose (YPD) agar plates (2% (w/v) peptone, 1% (w/v) yeast extract, 2% (w/v) glucose, and 2% (w/v) agar) at 30°C and then stored as frozen stocks in physiological saline supplemented with 20% (v/v) glycerol at −80°C. The test strains were activated by being first incubated on YPD agar plates for 24 hr at 30°C. The cells were then inoculated into YPD broth and grew overnight at 30°C and 200 rpm.
Saccharomyces cerevisiae YPH499 strain and C. albicans CAI4 strain were selected for genetic modification. E. coli strain DH5α was used for plasmid construction based on a previous method. Plasmids pESC-URA, URA3-Clox, pEXP, and pUG6 were used to construct expression vectors.12,13
Agents
FLC and KTC were purchased from the National Institute for the Control of Pharmaceutical Biological Products. ITC was obtained from Xian-Janssen Pharmaceutical Co., Ltd.. VRC was from TCI Co., Ltd.. POS was from Maya Reagent Co., Ltd.. Geldanamycin (GdA), radicicol (RAD), cyclosporin A (CsA), and tacrolimus (FK506) were purchased from Sigma. All the compounds were dissolved in dimethyl sulfoxide (DMSO) to obtain a stock solution of 10 mg/mL and then stored at −20°C. Final desired concentration was diluted with RPMI 1640 medium.
Antifungal susceptibility tests
The minimum inhibitory concentration (MIC) values of FLC, KTC, ITC, VRC, and POS against 22 C. albicans isolates were determined using the broth microdilution method according to the Clinical and Laboratory Standards Institute (CLSI) M27-A3 standard guidelines. 14 C. albicans SC5314 was used as control. Briefly, RPMI 1640 medium buffered with 0.165 M 3-N-morpholinepropanesulfonic acid (MOPS), pH 7.0, was used for the broth microdilution test. Twofold dilutions of the drugs were performed and distributed in 96-well flat bottom plates. The tested concentration of FLC ranged from 0.5 to 128 μg/mL, while those for KTC, ITC, VRC, and POS ranged from 0.03 to 16 μg/mL. The fungal inoculum was prepared from a 24-hr YPD broth culture incubated at 30°C; the cells were harvested in RPMI medium and diluted to about 0.5–2.5 × 103 cells/mL. The plates were incubated at 35°C for 24 hr. The MICs of the drugs were determined according to the CLSI M60 recommendations. 15 For FLC, the values of ≤2, 4, and ≥8 μg/mL correspond to susceptible (S), susceptible dose dependent, and resistant (R), respectively. For VRC, these values are ≤0.12 μg/mL (S), 0.25–0.5 μg/mL (Intermediate, I), and ≥1 μg/mL (R). Clinical breakpoints for POS, ITC and KTC have not been proposed yet.
Checkerboard assay of combined antifungal activity
The interaction of FLC and Hsp90 or calcineurin inhibitors against the four resistant and two sensitive isolates was assessed using the broth microdilution checkerboard assay, as previously described.16,17 Briefly, the culture was diluted to a final concentration of 0.5–2.5 × 103 cells/mL with RPMI 1640 medium. In a 96-well plate, a fixed concentration of Hsp90 or calcineurin inhibitors and twofold dilutions of FLC were mixed for a total volume of 100 μL per well. The concentration of Hsp90 inhibitors GdA and RAD was 2.5 μg/mL. The concentration of calcineurin inhibitors CsA and FK506 was 5 μg/mL. The FLC concentration gradient ranged from 0.0625 to 4 μg/mL. The plates were incubated at 35°C and the growth was monitored by recording the OD595 values using a BioTek Synergy H1 Microplate Reader.
Flow cytometric analysis of the efflux of rhodamine 6G
The activity of efflux pumps in isolates 28A, 28D, 28I, and 28J was evaluated using flow cytometry by measuring the efflux of rhodamine 6G as previously described. 18 Activated C. albicans cells (106) were suspended in 2 mL of phosphate buffer saline (PBS) and incubated at 30°C with constant shaking for 4 hr to induce starvation. Rhodamine 6G at 10 μM was then added and incubated for another 30 min at 30°C. The cells were harvested and washed twice in cold sterile PBS and then resuspended. The fluorescence was immediately quantified using a FACScan flow cytometer (FACSCalibur; BD). Glucose (1%) was then added and incubated at 30°C for 30 min. The fluorescence of 105 cells was then evaluated by the flow cytometer. The fluorescence of the cells incubated without rhodamine 6G served as an unstained control. The fluorescence was expressed as geometric mean (Gmean) values and the data presented corresponded to fluorescence frequency distribution histograms. Each experiment was performed in triplicate.
Quantitative real-time polymerase chain reaction
C. albicans isolates were grown to the exponential phase in the YPD medium and total RNAs were extracted using the hot phenol method as previously described, and converted to complementary DNA (cDNA) using a cDNA synthesis kit (TaKaRa Biotechnology, Dalian, China). 19 The polymerase chain reaction (PCR) reactions were conducted using a SYBR Green master mix (TaKaRa Biotechnology) in an Eppendorf Mastercycler real-time PCR system. The following parameters were used for the real time-PCR amplification: heat inactivation of reverse transcriptase at 95°C for 2 min; and then 40 PCR cycles (95°C for 20 sec, 60°C for 20 sec, and 72°C for 10 sec). The primers used in real-time PCR reactions are listed in Supplementary Table S1. 18S ribosomal RNA (rRNA) served as the internal control. 20 The results are represented as the mean ± standard deviation (SD).
PCR amplification and gene sequencing
Genomic DNA was extracted from different C. albicans strains as described by Versalovic et al., and used to amplify full-length sequences of ERG11, ERG3, UPC2, MRR1, and CAP1 genes. 21 The primers used are listed in Supplementary Table S2. The PCR products were sequenced by Biosune, Inc. (Shanghai, China) and compared with the sequences published in the GenBank (ERG11, AY856352; ERG3, AF069752; UPC2, EU583453; MRR1, EU497768; and CAP1, U95611). We submitted the new sequences obtained in this study to the GenBank and the accession numbers are as follows: CAP128A, MW658177; CAP128D, MW658178; ERG328A, MW658179; ERG328D, MW658180; ERG328I, MW658181; ERG1128D-1, MW658182; and ERG1128D-2, MW658183.
Construction of C. albicans strains harboring mutated alleles of CAP1 or ERG3
To mimic the heterozygosity of strains 28A, 28D, and 28I, one copy of CAP1 or ERG3 in C. albicans CAI4 was disrupted using the plasmids URA3-Clox, as previously described. 22 The mutated CAP1 alleles of 28A and 28D, ERG3 alleles of 28A, 28D, and 28I, and synthesized ERG3 sequences with single mutation of N324S were cloned into the plasmid pEXP under the control of MET3 promoter, and inserted into PRS1 locus through homologous recombination to obtain strains CAI4-CAP128A, CAI4-CAP128D, CAI4-ERG328A, CAI4-ERG328D, CAI4-ERG328I, and CAI4-ERG3N324S, respectively. Transformation with the wild-type copy of CAP1 or ERG3 of SC5314 yielded the strains CAI4-CAP1wild and CAI4-ERG3wild as control in susceptibility test. The susceptibility tests of the constructed strains to azole drugs were then performed. The primers used in the construction of these strains are listed in Supplementary Table S3.
Expression of CaERG11 in S. cerevisiae
The ERG11 ORFs of the wild-type strain SC5314 and azole-resistant strain 28D with two different mutated alleles ERG11–1 and ERG11–2 were amplified using primers containing restriction sites for the BamHI and XhoI enzymes (Supplementary Table S3). The amplified BamHI-XhoI fragment was then ligated into the BamHI-XhoI sites of pESC-URA, which contains the GAL1 promoter and CYC1 terminator sequences. Because S. cerevisiae cannot survive in the absence of a functional lanosterol 14α-demethylase, S. cerevisiae YPH 499 was first transformed with the construct plasmid using the Frozen-EZ Yeast Transformation II Kit (Zymo Research, Irvine, CA), and grown on the selective synthetic dropout-uracil plates (SD-URA) containing galactose (2% w/v) to induce the expression of ERG11. The selected colonies were checked by plasmid extraction and enzyme digestion. The genomic ERG11 of the constructed strain was then disrupted as previously described with some modifications. 23 Briefly, a linear kanamycin/G418 DNA fragment was amplified from the pUG6 plasmid using the ERG-G418 primers (Supplementary Table S3), and used to transform the corrected transformants mentioned above. S. cerevisiae transformants were then selected on YPD agar plates containing 200 μg/mL of geneticin (G418 sulfate; Sigma), and checked through PCR. Introduction of wild-type copy of ERG11 and mutated copies of ERG11–1 and ERG11–2 yielded the heterozygote mutants YPH 499-ERG11wild, YPH 499-ERG1128D1, and YPH 499-ERG1128D2, respectively.
Homology modeling of CaErg11p
The homology of CaErg11p was modeled using a SWISS-MODEL server with the crystal structure of lanosterol 14α-demethylase of S. cerevisiae with an intact transmembrane domain bound to ITC (PDB code: 5EQB) as the template.24,25
Statistical analysis
All data were expressed as the mean ± SD. Statistical significance between the treatment and control groups was analyzed by using the Student's t test (two tailed, unequal variance). A p-value below 0.05 was considered to indicate statistical significance.
Results
The azole-resistant strains rely on Hsp90 to maintain their azole resistance
We totally obtained 22 clinical C. albicans isolates from 7 patients at the affiliated teaching hospitals of Shandong University in Jinan. The susceptibility profiles of these strains to five different azole drugs (FLC, KTC, ITC, VRC, and POS) were tested according to the CLSI M27-A3 standard guidelines. Wild-type strain SC5314 was selected as control. The MIC values of FLC, KTC, ITC, VRC, and POS against different isolates are summarized in Table 1. Four isolates (28A, 28D, 28I, and 28J) exhibited distinct high-level resistance to all tested azoles (MIC >128 μg/mL for FLC and MIC >16 μg/mL for KTC, ITC, VRC, and POS). The susceptibility profiles of isolates 28A, 28D, 28I, and 28J at 30°C and 37°C were also determined. The results have shown that all the strains exhibit stable pan-azole resistance that is independent of temperature (data not shown).
The Minimum Inhibitory Concentration Values and the Expressions of Efflux Pump Encoding Genes in 22 Clinical Candida albicans Isolates
For azole-resistant isolates, genes with a measured ΔCT value that fell outside the 3-SD range are in bold.
—, Nontested; Avg ΔCT, the average ΔCT values for susceptible strains and SC5314; FLC, fluconazole; ITC, itraconazole; KTC, ketoconazole; MIC, minimum inhibitory concentration; POS, posaconazole; SD, standard deviation; VRC, voriconazole.
Hsp90 is an essential molecular chaperone that plays a key role in regulating survival, virulence, and drug resistance in diverse pathogens. 26 The checkerboard assay combining Hsp90 inhibitors and FLC revealed that both GdA and RAD abolished FLC resistance of these azole-resistant strains (Fig. 1), suggesting that their azole resistance was dependent on the proper function of Hsp90.
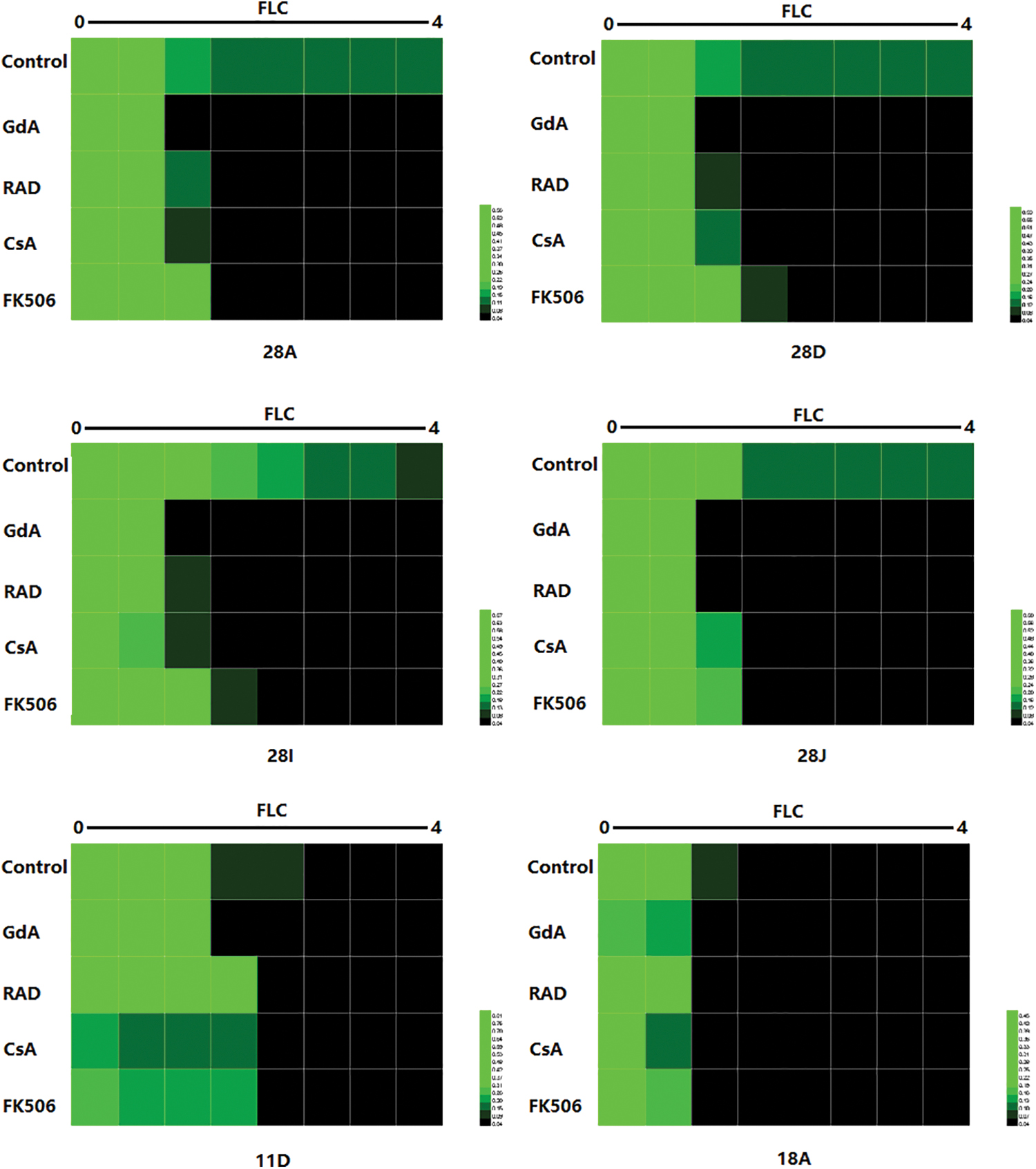
Hsp90 regulates azole resistance in a subset of clinical isolates. The interactions of FLC and Hsp90 or calcineurin inhibitor activities against azole-resistant and azole-sensitive isolates were determined in a checkerboard assay. The assays were performed in RPMI 1640 medium at 35°C. Cell growth of the indicated strains was monitored based on OD595 readings after 48 hr in the presence of the indicated drugs. HemI (version 1.0) was used to figure out the results. FLC, fluconazole. Color images are available online.
Calcineurin, the client protein of Hsp90, is reported to be essential for the development of azole resistance in C. albicans and mediates the Hsp90-dependent azole resistance. Compromising the calcineurin function genetically or pharmacologically enhances the susceptibility of C. albicans to azoles. 27 The inhibitory effect of CsA and FK506, the inhibitors of calcineurin, on the growth of these azole-resistant strains was tested individually or in combination with FLC. At 5 μg/mL, neither CsA nor FK506 alone inhibited the growth of the resistant strains; however, they exhibited inhibitory effect in combination with FLC (Fig. 1). This suggested that compromising the calcineurin function phenocopied the effect of Hsp90 inhibition and reversed azole resistance in the tested strains. A synergistic effect was also observed between FLC and Hsp90 inhibitors/calcineurin inhibitors in azole-susceptible strains 11D and 18A, which is consistent with previous findings.
ERG3 mutations in isolate 28I contribute to its azole resistance
ERG3 encodes a key enzyme sterol Δ5, 6-desaturase in the late stage of ergosterol synthesis. Its mutations can block the generation of the toxic metabolites when Erg11 is inhibited by azole antifungals, and therefore induce occurrence of azole resistance. 28 Gene sequencing of ERG3 from the four Hsp90-dependent resistant isolates 28A, 28D, 28I, and 28J revealed four amino acid substitutions (S265Y, N322D, N324S, and E355D) in Erg3 for the first time (Table 2). To determine the role of these substitutions in azole resistance development, C. albicans strains CAI4 with one copy of the mutated ERG3 were constructed, mimicking the heterozygosity of ERG3 in the 28A, 28D, and 28I strains. The azole susceptibility of the constructed C. albicans strains was then tested. The MIC value of FLC against C. albicans CAI4-ERG328I increased fourfold compared with the wild-type C. albicans strain (from 2 to 8 μg/mL), whereas CAI4-ERG328D and CAI4-ERG328A did not acquire any azole resistance (Table 3). In addition, the sensitivity of constructed CAI4-ERG328I strain to other azole agents also decreased, suggesting that the combination of substitutions of S265Y, N322D, and N324S in Erg3 plays an important role in mediating azole resistance in isolate 28I. The heterozygous strain containing single mutation (N324S) in Erg3 in CAI4 strain did not exhibit great resistance to azole drugs, suggesting that N324S mutation alone played a minor role in azole resistance occurrence.
Missense Mutations in Related Genes of Resistant Isolates of Candida albicans
All mutations are heterozygous (mutation in a single allele).
The amino acid substitutions in genes that are unreported are shown in bold font.
—, nontested; N.D., nondetected.
Minimum Inhibitory Concentrations of Fluconazole Against Candida albicans Isolates with Mutated Allele of CAP1, ERG3, and ERG11
The amino acid substitutions contributing to azole resistance in 28I are shown in bold.
Increased efflux activity and upregulated MDR1 expression observed in isolates 28A and 28D
To determine the role of efflux-mediated resistance in the evaluated set of clinical isolates, the activity of efflux pumps and the messenger RNA (mRNA) transcript levels of the multidrug transporter-encoding genes were evaluated. The activity of efflux pumps was assessed using flow cytometry by measuring the efflux of rhodamine 6G. The intracellular uptake of rhodamine 6G in azole-resistant strains and azole-susceptible strains was similar. However, rhodamine 6G efflux from azole-resistant strains 28A and 28D was greater than that in azole-susceptible strains in the presence of glucose (Fig. 2A). Relative geometric mean values (ΔGmean) of tested strains were calculated as ΔGmean = Gmean [uptake] − Gmean [efflux]. As shown in Fig. 2B, the ΔGmean values for 28A and 28D increased by 5.03- or 6.26-fold compared with the average ΔGmean values for susceptible strains and SC5314, indicating high efflux activity.

Flow cytometry analysis of rhodamine 6G uptake and efflux in four azole-resistant strains with azole-susceptible strains as control.
We next assessed the transcriptional levels of CDR1, CDR2, and MDR1 in strains 28A and 28D. Genes with a measured ΔCT value that fell outside the 3-SD range of that measured in the azole-susceptible C. albicans isolates were designated as being overexpressed or underexpressed. High level of MDR1 expression was observed in both 28A and 28D, which have upregulated expression of MDR1 by 2.63- and 2.93-fold, calculated according to the formula 2−ΔΔCT, respectively. However, we did not observe the upregulation of CDR1 and CDR2 in either strain (Table 1 and Supplementary Fig. S1).
MRR1 and CAP1, which encode the transcription factors controlling the expression of multidrug efflux pumps in C. albicans, were also amplified and sequenced from the isolates 28A and 28D, to determine the basis of MDR1 overexpression in these strains. 29 No mutations in the MRR1 gene was detected in these two tested strains. Four new heterozygous mutations underpinning the amino acid substitutions of K65R, M140I, V200M, and M308T were identified in CAP1 in the azole-resistant strains 28A and 28D (Table 2). To determine the role of these mutations in azole resistance, we introduced mutated CAP1 into C. albicans CAI4 with one copy of CAP1 disrupted. The azole susceptibility of the constructed C. albicans strains was tested. The MIC values of azole antifungals against C. albicans CAI4-CAP128A and CAI4-CAP128D were close to that of wild-type strain (Table 3), suggesting that these mutations in CAP1 contribute little to azole resistance.
Real-time PCR reveals overexpression of ERG11 in isolate 28J
The expressions of ERG11 in these isolates were measured using quantitative real-time PCR and the strain 28J displayed significant upregulation in ERG11 compared with azole-susceptible strains (Fig. 3). Upc2 is a C. albicans zinc cluster transcription factor that regulates the expression of genes involved in sterol biosynthesis and the uptake of sterol. A gain-of-function mutation in UPC2 that leads to increased expression of ERG11 and contributes to azole resistance in C. albicans has been reported previously. 30 Therefore, the open reading frame (ORF) of UPC2 in 28J was also sequenced, while no mutation was found. Structural alterations of the protein Erg11 caused by gene mutations are one of the most common mechanisms underlying the azole resistance. 31 DNA sequence analysis of ERG11 ORF revealed six amino acid substitutions in the azole-resistant isolate 28D (Table 2). I162S and D116E were present in one allele, while V185A, P390L, P230L, and I261V mutations occurred in the other one. Among these, D116E is not associated with azole resistance as reported in previous study. 32 The role of I261V in azole resistance has not been clearly addressed, although this substitution was indeed detected in an azole-resistant isolate. 33 The P230L substitution alone is not sufficient to disrupt its binding with POS, and the presence of additional substitutions is required to cause changes in the MIC of POS. 34 Three other mutations (I162S, V185A, and P390L) were novel mutations. As shown in a three-dimensional (3D) model of the target enzyme Erg11 interacting with an azole (Fig. 4), the three novel substitutions were located either in the α-helix (I162S) or β-pleated sheets (V185A and P390L). None of them was in the vicinity of the active center or the substrate access channel of the protein, suggesting little possibility of these mutations in conferring azole resistance. To confirm this prediction, the full-length ERG11 ORF amplified from C. albicans 28D or wild-type strain SC5314 was cloned into plasmid pESC-URA and transformed into S. cerevisiae YPH 499. The obtained strains were tested for azole susceptibility. As illustrated in Table 3, the MIC of FLC against YPH 499-ERG1128D1 and YPH 499-ERG1128D2 strains increased by twofold compared with that of YPH 499-ERG11wild, but no increase of MIC values was observed among these strains when tested using other azole drugs. This suggested that these new substitutions had minor contributions to azole resistance.
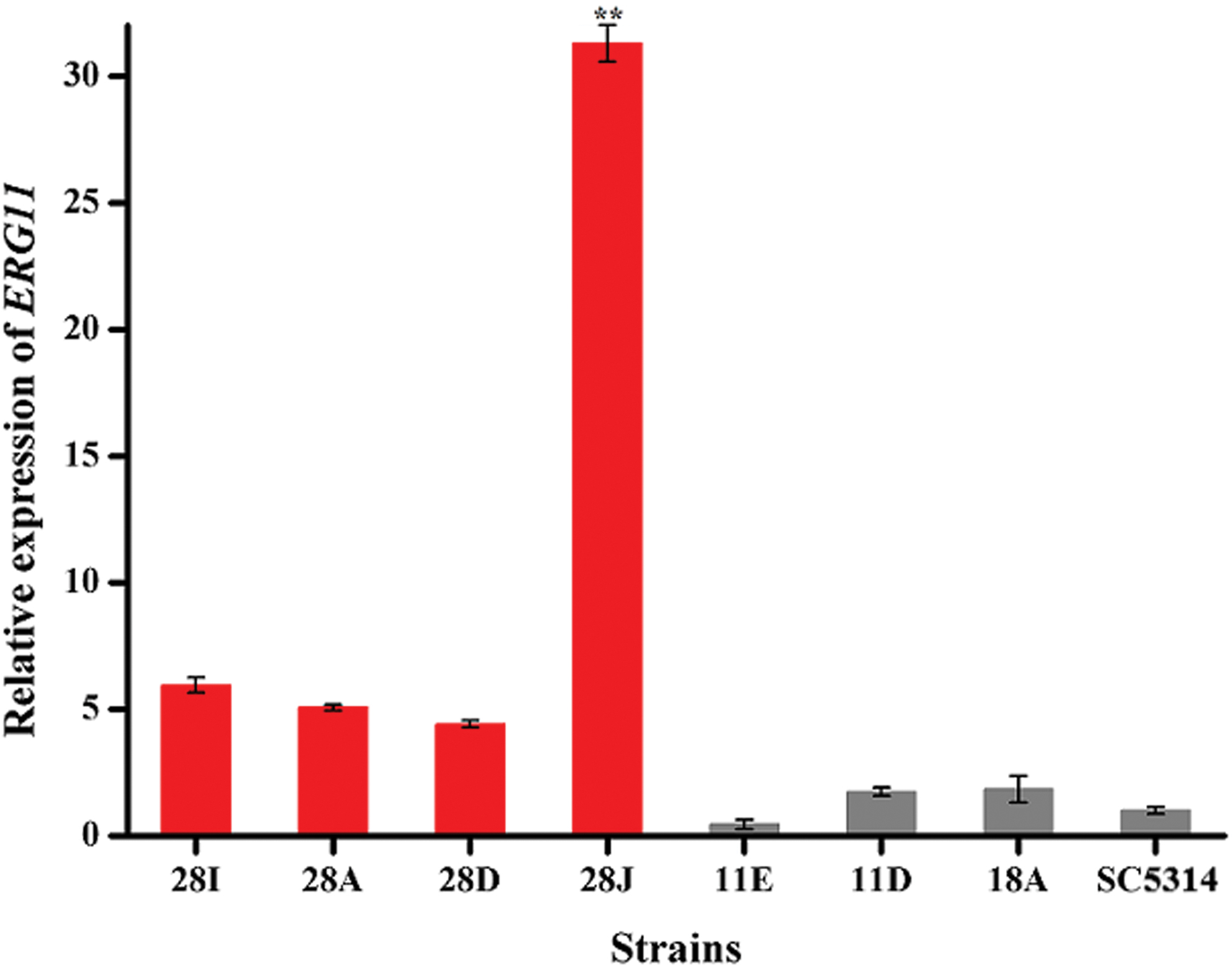
The expressional levels of ERG11 in azole-susceptible (gray bars) and azole-resistant (red bars) isolates of Candida albicans. Four azole-resistant strains (28A, 28D, 28I, and 28J) and four azole-susceptible strains were grown in YPD medium. Transcriptional levels of ERG11 gene were indicated as a fold change relative to that of the housekeeping gene 18S rRNA using qPCR assay. Each sample was processed in triplicate. Bars represent mean ± SD. **p < 0.01 versus susceptible strains. qPCR, quantitative polymerase chain reaction; rRNA, ribosomal RNA; YPD, yeast–peptone–dextrose. Color images are available online.

Homology model of CaErg11p structure. The homology of CaErg11p was modeled with the crystal structure of lanosterol 14α-demethylase of Saccharomyces cerevisiae bound to ITC. α-helices and β-pleated sheets are shown in blue and purple, respectively. The azole compound was shown in ball-and-stick model. The substitutions were highlighted in bold. ITC, itraconazole. Color images are available online.
Discussion
Azole-resistant strains were found frequently in cancer patients who have received chemotherapy and antifungal treatment, and hindered the efficient control of serious fungal infection. 35 In this study, we identified four pan-azole-resistant isolates in 22 clinical C. albicans strains collected from cancer patients. We found that compromising Hsp90 function or its downstream effectors by the specific inhibitors was able to abrogate the FLC resistance in all these strains. Design and synthesis of fungal-selective Hsp90 inhibitors with lower toxicities is a promising strategy to cripple clinical resistant fungal pathogens.36,37 The mechanisms by which Hsp90 mediates the evolution of fungal drug resistance involve the induction of gene mutations or overexpression.38,39 Previous research have shown that 12 Hsp90-dependent resistant S. cerevisiae strains isolated by rapid selection contained ERG3 mutations. 40 ERG3 mutations also occurred frequently in clinical azole-resistant C. albicans and were regarded as potential causes of their azole resistance. Amino acid substitutions of V237A, H243N, A255T, E266D, T330A, W332R, A351V, and A353T in Erg3 have been documented previously; however, their roles in resistance have not been fully proved.41,42 In this study, we have confirmed four novel ERG3 mutations in the isolates 28A, 28D, and 28I and successfully established C. albicans CAI4 constructs with copies of the mutated ERG3. It turns out that the substitution (S265Y, N322D, and N324S) combination in Erg3 identified in strain 28I was able to increase FLC resistance of the construct by fourfold. However, other mutations in ERG3, such as S265Y and E355D in strain 28D and S265Y and N322D in strain 28A, did not confer FLC resistance to the modified strain. The results showed that not all mutations in ERG3 can definitely cause the changes in protein function, and then lead to drug resistance. It is necessary to select a suitable model to verify the function of the variations. Besides, amino acid substitutions detected in Erg11 in strain 28D have minor contributions to its azole resistance. Currently, no 3D structural information of Erg3 protein is available yet. As shown above, the crystal structure of Erg11 provides a potential reliable model for the evaluation of the role for the amino acids at different positions in maintaining the activity of the enzyme. This reminds us that the resistance-related amino acid substitutions in Erg3 in clinical resistant isolates might interact with each other and form an active center and substrate channel in the protein. Identification of specific sites that are responsible for its activity may provide useful information for further structural studies and drug design.
Increased expression of drug efflux pumps and Erg11, as well as its mutations are three well-described mechanisms that contribute to azole resistance. A combination of the mechanisms is usually necessary to confer clinically relevant levels of azole resistance. 43 In this research, we found that significant overexpression of ERG11, the pharmacological target of azoles, occurred in isolate 28J. The finding was that no mutation in the gene UPC2 occurred, suggesting that other ERG11 regulators in this strain exist and need to be further investigated in the future study. Increased activity of efflux pumps together with overexpression of MDR1 were observed in isolates 28A and 28D, implying that long-term treatment with azoles may result in drug resistance by increasing the levels of expression and the activities of efflux pumps. However, overexpression of CDR1 and CDR2 was not observed in any isolate. These results demonstrate that overexpression of multidrug resistance genes could induce drug resistance in C. albicans, but overexpression of the above three genes did not definitely occur at the same time. Regulation of MDR1 expression by zinc cluster proteins, such as transcription factors Mrr1 and Cap1, is proved to be involved in the development of drug resistance. A C-terminally truncated CAP1ΔC333 allele caused MDR1 overexpression in a wild-type strain SC5314. 44 The mRNA levels of Cap1 and Mrr1 were significantly increased in the FLC-resistant strains collected from patients with vulvovaginal candidiasis compared with strains sensitive to the azole agents. Missense mutations of S381N, P311S, and A390T in Cap1, as well as T917M, T923I, N937K, E1020Q, F1032L, and S1037L in Mrr1 were identified in the related isolates. However, they did not test whether these missense mutations are associated with drug resistance in C. albicans strains. 45 In this study, we have also described four novel mutations of K65R, M140I, V200M, and M308T in Cap1 from isolates 28A and 28D and tested their impact on the susceptibility of C. albicans to azole drugs using the CAI4 model. Unfortunately, they did not influence the susceptibility of C. albicans strains to multiple drugs, suggesting that other regulators or signal pathways involved in MDR1 overexpression need to be further investigated.
Great heterogeneity among the C. albicans colonies isolated from the same host was observed in both phenotypic and genotypic aspects. 46 FLC-resistant and FLC-sensitive strains were detected within isolates recovered from the same cancer patient, making the choice of treatment strategy of fungal infections even more difficult. Isolates 28A, 28D, 28I, and 28J are four azole-resistant colonies isolated from the same patient, having the same level of MIC. However, distinct resistance mechanisms were identified to be responsible in the four strains in our research. The results indicated that diverse molecular mechanisms, such as changes in gene expression levels, mutations in protein sequence, and modifications in related signaling pathways in C. albicans strains, combined to contribute to their heterogeneity in azole susceptibility. Comprehensive characterization of strain specimen collected from patients might be useful to provide a more reliable assessment and avoid the aberration application of antifungals in clinical practice.
In this study, we found that the mechanism of developing resistance to azoles in C. albicans even collected from the same patients is a complex process and no one single factor alone can cause azole resistance. Hence, combination therapy that targets different mechanisms should be adopted in clinical treatment of candidiasis. Our study deepens the understanding of the local determinants of azole resistance, and may lead to further insights that will aid the development of novel targeted treatments of C. albicans infections.
Footnotes
Acknowledgments
We thank Prof. Qingguo Qi in Shandong University for donating the clinical C. albicans strains and Professor Alistair J.P. Brown in University of Aberdeen of United Kingdom for the plasmid gifts used for gene deletions.
Authors' Contributions
H.S., Y.Z., W.C., and M.Z. performed the experiments. H.S., Y.Z., W.C., and H.L. designed the research. H.S., Y.Z., W.C., and H.L. analyzed the data and wrote the article. All authors approved the article for publication.
Disclosure Statement
The authors declare that they have no conflict of interests.
Funding Information
This work was funded by National Natural Science Foundations (Nos. 81773786 and 81630093), Natural Science Fund for Excellent Young Scholars of Shandong Province of China (ZR2020YQ63), and Young Scholars Program of Shandong University (2017WLJH41).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.