
Abstract
Proteus vulgaris is an important foodborne opportunistic pathogen, both environmentally and clinically. The use of appropriate antibiotics has significant therapeutic effects, but has led to the emergence and spread of drug-resistant strains. In this study, a P. vulgaris strain, designated “P3M,” was isolated from Penaeus vannamei in Tianjin, China. The whole genome of P3M was sequenced, generating detailed information, including the key genes involved in important metabolic pathways and their physiological functions. A total of 218 antibiotic resistance genes (ARGs) were predicted in the genome. The determination of various minimum inhibitory concentrations indicated that P3M is a multidrug-resistant (MDR) bacterium, with significant resistance to 16 antibiotics in seven categories. Determination of fractional inhibitory concentration index showed that the combination of ciprofloxacin plus tetracycline exhibited synergistic antimicrobial activity. Bioinformatics and phylogenetic analyses detected the presence of two two-component systems that mediate multidrug resistance and several mobile genetic elements involved in the horizontal transfer of ARGs in P3M. P. vulgaris strains represent a serious challenge to clinicians and infection control teams for its ubiquity worldwide and close relevance with human life. To the best of our knowledge, we report the first isolation and characterization of an important foodborne MDR P. vulgaris strain, and this study will provide necessary theoretical basis for the selection and clinical use of the appropriate antibiotics.
Introduction
The Proteus, a genus in the family Enterobacteriaceae, is one of the commonest Gram-negative genera of clinical opportunistic pathogens and is widely distributed in water, soil, contaminated food, human and animal intestines, usually causing urinary tract infections, wound and burn infections, and respiratory tract infections.1,2 Proteus contains the species Proteus mirabilis, Proteus vulgaris, Proteus penneri, Proteus hauseri, Proteus cibarius, Proteus terrae, and three unnamed genomospecies (genomospecies 4, 5, and 6).1,3–6 Of these, P. vulgaris and P. mirabilis are closely associated with clinical diseases, posing a serious threat to human health as they circulate through food chains and ecosystems.1,7,8 In clinical practice, a large number of antibiotics are used to treat the infections caused by P. vulgaris and P. mirabilis, which has led to the emergence and transmission of antibiotic resistance in the environment.9–12
The emergence of multidrug-resistant (MDR) bacteria is one of the top three threats to global public health today, and is largely caused by the overuse of drugs and the inappropriate consumption and application of antibiotics.13–15 In recent years, the isolation rate of resistant Proteus spp. in clinical specimens has gradually increased, and some are MDR bacteria, which dramatically reduces the therapeutic options.12,16–18 Polymyxin-resistant bacteria are also gradually entering the field of view, although this kind of antibiotic has been described as the last line of therapeutic defense against infections caused by MDR Enterobacteriaceae, undoubtedly increasing the difficulty of clinical treatment even further.19–21 MDR bacteria thrive in harsh natural environments by acquiring various preexisting antibiotic resistance genes (ARGs). Mobile genetic elements (MGEs; including plasmids, integrative and conjugative elements, transposons and insertion sequences, integrons, and bacteriophages) play crucial roles in the rapid evolution and environmental adaption of the bacteria through this process.22–29 MGEs make possible the intracellular and intercellular mobility of antimicrobial determinants, contributing greatly to the horizontal transfer of ARGs and the further spread of multidrug resistance.30,31
In this study, an MDR P. vulgaris strain, showing significant resistance to polymyxin, was isolated from the intestine of Penaeus vannamei in Tianjin, China. The possible transmission of ARGs, the basic characteristics and antimicrobial profile of P3M were analyzed in detail. This study clarifies the antibiotic resistance and transmission characteristics of the species P. vulgaris, and provides necessary fundamental references for the rational use of antibiotics in its clinical treatment.
Materials and Methods
Animal experiments complied with National and Chinese national guidelines for the use of animals in research. The protocol was approved by the Animal Care and Use Committee of the College of Life Sciences, Nankai University (permission number NK-04-2012).
Strain isolation and growth conditions
P. vulgaris strain P3M was isolated from the intestine of Pe. vannamei, which was purchased from a supermarket in Tianjin, China, and was screened for and cultured on Luria–Bertani (LB) agar containing 32 mg/L erythromycin at 37°C for 12 hours. P3M was identified with 16S ribosomal RNA (rRNA)-directed polymerase chain reaction and sequencing methods. 32
Whole-genome sequencing
A next-generation sequencing library was constructed following the manufacturer's protocol. The library with different indices was then multiplexed and loaded onto an Illumina HiSeq instrument according to manufacturer's instructions (Illumina, San Diego, CA). Sequencing was performed with a 2 × 150 paired-end (PE) configuration, and image analysis and base calling were performed with the HiSeq Control Software (HCS)+OLB+GAPipeline-1.6 (Illumina) on the HiSeq instrument.
For PacBio sequencing, the genomic DNA was sheared, and 10 kb (20 kb if the genome was larger than 30 Mb) double-stranded DNA fragments were selected. The DNA fragments were end repaired and ligated with universal hairpin adapters. The subsequent steps in preparing the SMRTbell library were according to the manufacturer's instructions. The library was sequenced with a PacBio RSII/Sequel Single Molecule Real Time instrument. 33 The PacBio reads were assembled with HGAP4/Falcon in WGS-Assembler 8.2.34–36 The genome was then corrected with the software Pilon using previous illumina data. The Prodigal/Augustus gene-finding software was used to identify coding genes.37,38 Transfer RNAs (tRNAs) were detected in the genome with the program tRNAscan-SE with the default parameters. 39 rRNAs were identified with RNAmmer. 40 Whole-genome sequencing (WGS) was performed and the results analyzed by GENEWIZ, Inc. (Suzhou, China).
Antimicrobial susceptibility testing
The minimum inhibitory concentrations (MICs) of P3M were determined with the broth microdilution method, according to the Clinical and Laboratory Standards Institute (CLSI) recommendations, 41 using an AST panel for aerobic Gram-negative bacilli (Biofosun, Shanghai, China) that included 28 antibiotics. The CLSI breakpoints criteria (S: susceptible; I: intermediately resistant; R: resistant) for these antibiotics were: cefazolin (≤2, 4, ≥8) mg/L (S, I, R); cefotaxime (≤1, 2, ≥4) mg/L (S, I, R); ceftazidime (≤4, 8, ≥16) mg/L (S, I, R); cefepime (≤2, 4–8, ≥16) mg/L (S, I, R); nalidixic acid (≤16, ≥32) mg/L (S, R); ciprofloxacin (CIP; ≤1, 2, ≥4) mg/L (S, I, R); levofloxacin (LEV; ≤2, 4, ≥8) mg/L (S, I, R); gemifloxacin (≤0.25, 0.5, ≥1) mg/L (S, I, R); ampicillin (≤8, 16, ≥32) mg/L (S, I, R); ampicillin/sulbactam (≤8/4, 16/8, ≥32/16) mg/L (S, I, R); amoxicillin–clavulanate (≤8/4, 16/8, ≥32/16) mg/L (S, I, R); aztreonam (≤4, 8, ≥16) mg/L (S, I, R); gentamicin (≤4, 8, ≥16) mg/L (S, I, R); amikacin (≤16, 32, ≥64) mg/L (S, I, R); kanamycin (≤16, 32, ≥64) mg/L (S, I, R); streptomycin (≤8, 16, ≥32) mg/L (S, I, R); tetracycline (TET; ≤4, 8, ≥16) mg/L (S, I, R); minocyline (≤4, 8, ≥16) mg/L (S, I, R); doxycycline (DOX; ≤4, 8, ≥16) mg/L (S, I, R); colistin E (≤2, ≥4) mg/L (S, R); polymyxin B (≤2, 4, ≥8) mg/L (S, I, R); sulfamethoxazole–trimethoprim (≤2/38, ≥4/76) mg/L (S, R); sulfisoxazole (≤256, ≥512) mg/L (S, R); imipenem (≤1, 2, ≥4) mg/L (S, I, R); meropenem (≤1, 2, ≥4) mg/L (S, I, R); azithromycin (≤16, ≥32) mg/L (S, R); cefoxitin (≤8, 16, ≥32) mg/L (S, I, R); and chloramphenicol (≤8, 16, ≥32) mg/L (S, I, R). 41 Escherichia coli strain ATCC25922 was used as the quality control strain.
Checkerboard susceptibility testing
The antimicrobial activities of CIP, TET, and CHL in different combinations against P3M were tested in triplicate by the checkerboard method. 42 In this experiment, the combinations analyzed included CIP plus TET, CIP plus CHL, and TET plus CHL. The tests were performed on 96-well microdilution plates containing LB broth added with different concentrations (multiple and MIC fractions) of the antimicrobials in combinations. The P3M inoculum was prepared in LB broth without antimicrobial agents and added to the final concentration test of 10−6 CFU/mL. For growth control, P3M inoculum of 10−6 CFU/mL was grown in LB broth without antibiotics. After incubation of 20 hours at 37°C, the fractional inhibitory concentration index (FICI) was determined using the following formula: FICI = FICIA + FICIB, where FICIA/B represented the MIC of drug A/B in combination divided by the MIC of drug A/B alone. The FICI values were interpreted as follows: synergy, FICI ≤0.5; indifference, 0.5 < FICI <4; and antagonism, FICI ≥4. 43
Phylogenetic analysis
Unrooted neighbor-joining trees of P. vulgaris strains were generated from the indicated aligned sequences with MEGA7.44,45
Prediction of ARGs
All the ARGs in the P3M genome were predicted and analyzed with the Comprehensive Antibiotic Resistance Database (CARD), which provides data, models, and algorithms for the molecular basis of antimicrobial resistance, including selected reference sequences and single nucleotide polymorphism information obtained from Antibiotic Resistance Ontology (ARO).46,47
Kyoto Encyclopedia of Genes and Genomes pathway annotation
The genes involved in different metabolic pathways and in important biological functions were annotated and analyzed with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, which is used to systematically analyze gene functions and genomic information, and to map the corresponding biological pathways according to different types of biological processes. 48 This annotation allows researchers to study the gene and expression information within a whole network.
Cluster of Orthologous Groups annotation
Proteins with different physiological functions were predicted and analyzed with the Cluster of Orthologous Groups (COG) database, which is based on the phylogenetic relationships of coding proteins in complete bacterial genomes. 49 Because each cluster in COG is composed of specific orthologs, protein sequences can be annotated into a cluster of orthologous proteins by alignment and their functions predicted accordingly.
Accession number
The complete sequence of the P3M genome has been deposited in the National Center for Biotechnology Information repository under accession number CP060211.
Results
Description and characterization of the P3M genome
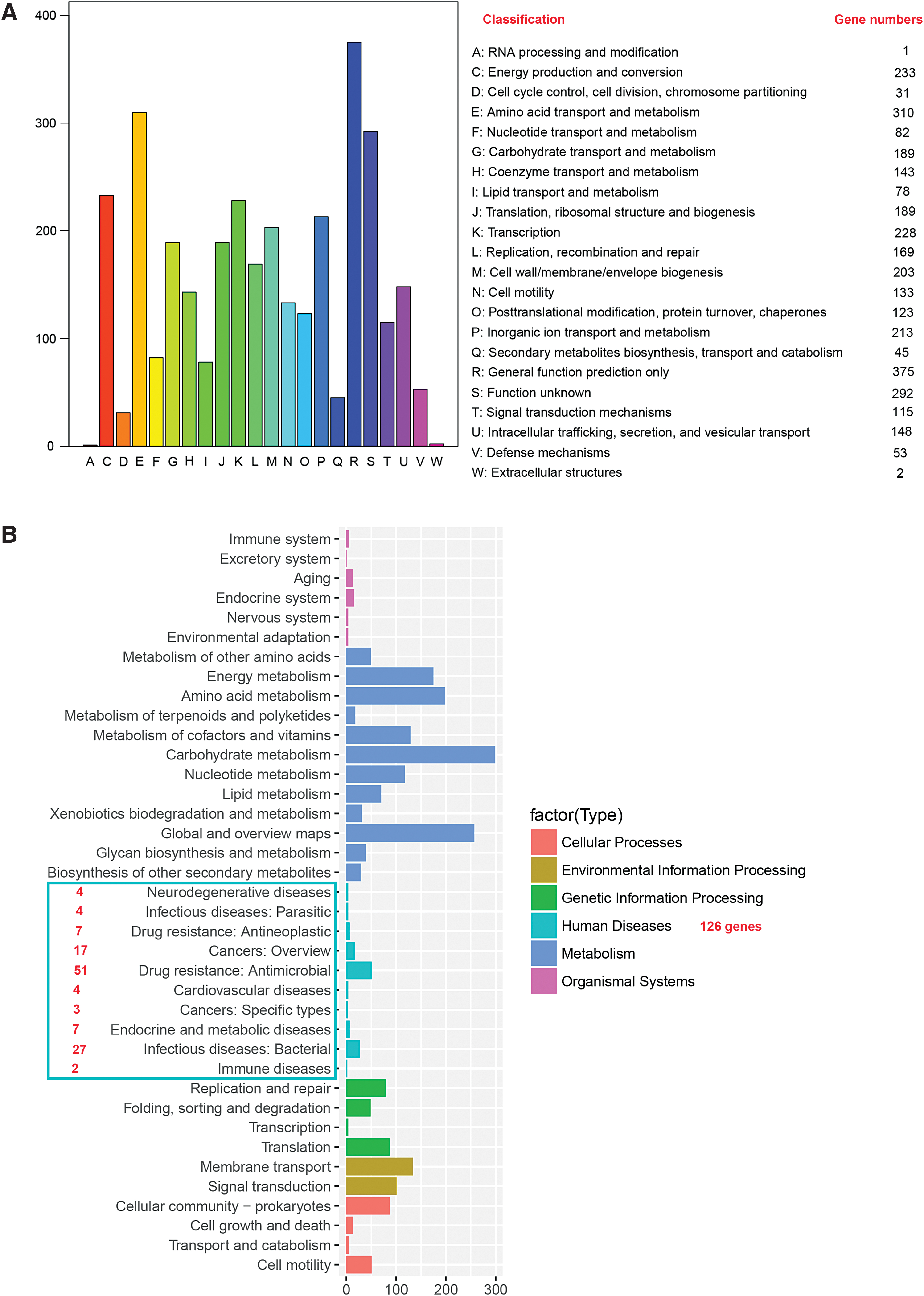
P3M was isolated from the intestine of Pe. vannamei in China and was initially identified as an erythromycin-resistant isolate. The P3M genome is 3,935,122 bp in size, with a guanine plus cytosine (GC) content of 37.97%. P3M contains two endogenous plasmids, p3M-2A (2,656 bp) and p3M-2B (5,903 bp), which have already been characterized. 50 A total of 3,613 coding genes were identified, including 3,502 coding sequences (CDSs) and 111 RNAs (Table 1). Of note, 218 ARGs were predicted in the genome, suggesting that P. vulgaris is not only an important opportunistic pathogen, but also a potential environmental reservoir for ARGs. The Circos (version 0.69) software was used to draw the genome circle map, 51 which provides detailed information about the gene distribution, noncoding RNAs (ncRNAs), GC content, and repeat sequences of P3M (Fig. 1).

Graphical circular representation of the genomic annotation information of P3M from circle 1 (outer) to circle 7 (inner). Circle 1, genome nucleotide numbering; circle 2, GC content; circle 3, genes encoded on the forward strand (red); circle 4, genes encoded on the reverse strand (green); circle 5, ncRNAs on the forward strand (blue); circle 6, ncRNAs on the reverse strand (purple); circle 7, information on the long repeat sequences in the genome (orange). GC, guanine plus cytosine; ncRNA, noncoding RNA. Color images are available online.
General Features of the P3M Genome
ARGs, antibiotic resistance genes; CDS, coding sequence; GC, guanine plus cytosine.
The functions of certain protein sequences were predicted and grouped into 22 functional classes by alignment with the COG database (Fig. 2A). The numbers of genes representing different functions in the genome vary greatly. For example, 310 genes are involved in amino acid transport and metabolism, whereas only 1 gene is predicted to be responsible for RNA processing and modification. These results allow us to further investigate the physiological metabolism and regulatory processes of P3M in the future. Many functional genes of P3M involved in different metabolic pathways were also identified in this study with a KEGG database analysis and were classified into six categories: cellular processes, environmental information processing, genetic information processing, human diseases, metabolism, and organismal systems. Each category was systematically divided into several secondary classifications, and the numbers of genes involved in the different biological metabolic pathways of the secondary classification were counted (Fig. 2B). The KEGG analysis showed that 126 genes in the genome of P3M are involved in metabolic pathways closely related to human diseases. It is noteworthy that nearly half of these genes (58 genes) are closely related to drug-resistance pathways: 7 of these genes are involved in antineoplastic-related pathways and 51 genes are closely related to antimicrobial.
Antimicrobial susceptibility profile of P3M
All the 218 genes related to antibiotic resistance in the genome of P3M were analyzed and classified into 36 specific categories according to the different types of resistance (Supplementary Fig. S1). Among them, the fluoroquinolone-resistance genes accounted for the largest proportion (19.72%). The smallest proportion of resistance genes (0.46%) was represented by bicyclomycin-, para-aminosalicylic acid-, sulfone-, antibacterial free fatty acids- and pyrazinamide-resistance genes (Supplementary Fig. S1 and Supplementary Table S1). Our analysis showed that among these 218 resistance genes, 155 were predicted to confer single resistance and 62 to confer multiple resistance, and 1 gene was associated with specific unknown resistance.
The MICs of several common classes of antibiotics against P3M were measured to determine its antibiotic resistance. As given in Table 2, P3M was sensitive to only 6 of the 28 selected antibiotics according to the CLSI breakpoint criteria: CIP (1 mg/L), LEV (2 mg/L), TET (4 mg/L), minocycline (MIN; 4 mg/L), DOX (4 mg/L), and CHL (4 mg/L). P3M was resistant to 16 antibiotics and intermediately resistant to the remaining 6 antibiotics. Therefore, based on the MIC and ARG analyses (Table 2 and Supplementary Fig. S1, respectively), we identified P3M as an MDR P. vulgaris strain.
Susceptibility Profile of P3M
All MICs were determined with broth microdilution assays, according to CLSI standards.
AMC, amoxicillin–clavulanate; AMI, amikacin; AMP, ampicillin; AMS, ampicillin/sulbactam; AZI, azithromycin; AZM, aztreonam; CAZ, ceftazidime; CFX, cefoxitin; CFZ, cefazolin; CHL, chloramphenicol; CIP, ciprofloxacin; CLSI, Clinical and Laboratory Standards Institute; CT, colistin E; CTX, cefotaxime; DOX, doxycycline; FEP, cefepime; GEM, gemifloxacin; GEN, gentamycin; I, intermediately resistant; IMI, imipenem; KAN, kanamycin; LEV, levofloxacin; MEM, meropenem; MIC, minimum inhibitory concentration; MIN, minocycline; NAL, nalidixic acid; PB, polymyxin B; R, resistant; S, susceptible; STR, streptomycin; SUL, sulfisoxazole; SXT, sulfamethoxazole–trimethoprim; TET, tetracycline.
Evaluation of combination antimicrobial activities
Combination therapy are now cumulatively becoming an essential approach in the treatment of MDR bacteria, in which two or more antibiotics are often used together to treat an infection against which one or both of the drugs may be ineffective alone. 52 In this study, we selected three representative antibiotics to which P3M is very sensitive according the antimicrobial susceptibility profile of P3M (Table 2): CIP, TET, and CHL, and determined their combined antimicrobial activities. As given in Table 3, the combinations of CIP with TET showed synergistic activity, presenting a significant reduction in MIC value of both antibiotics. However, the other two combinations did not show synergistic bacteriostatic effects, although the MIC of each antibiotic to P3M decreased to varying degrees.
Fractional Inhibitory Concentration Index Determined by Checkerboard Susceptibility Testing of P3M
FICI ≤0.5: S; 0.5 < FICI <4.0: I.
FICI, fractional inhibitory concentration index; i, indifferent; s, synergism.
Phylogenetic analysis of species P. vulgaris
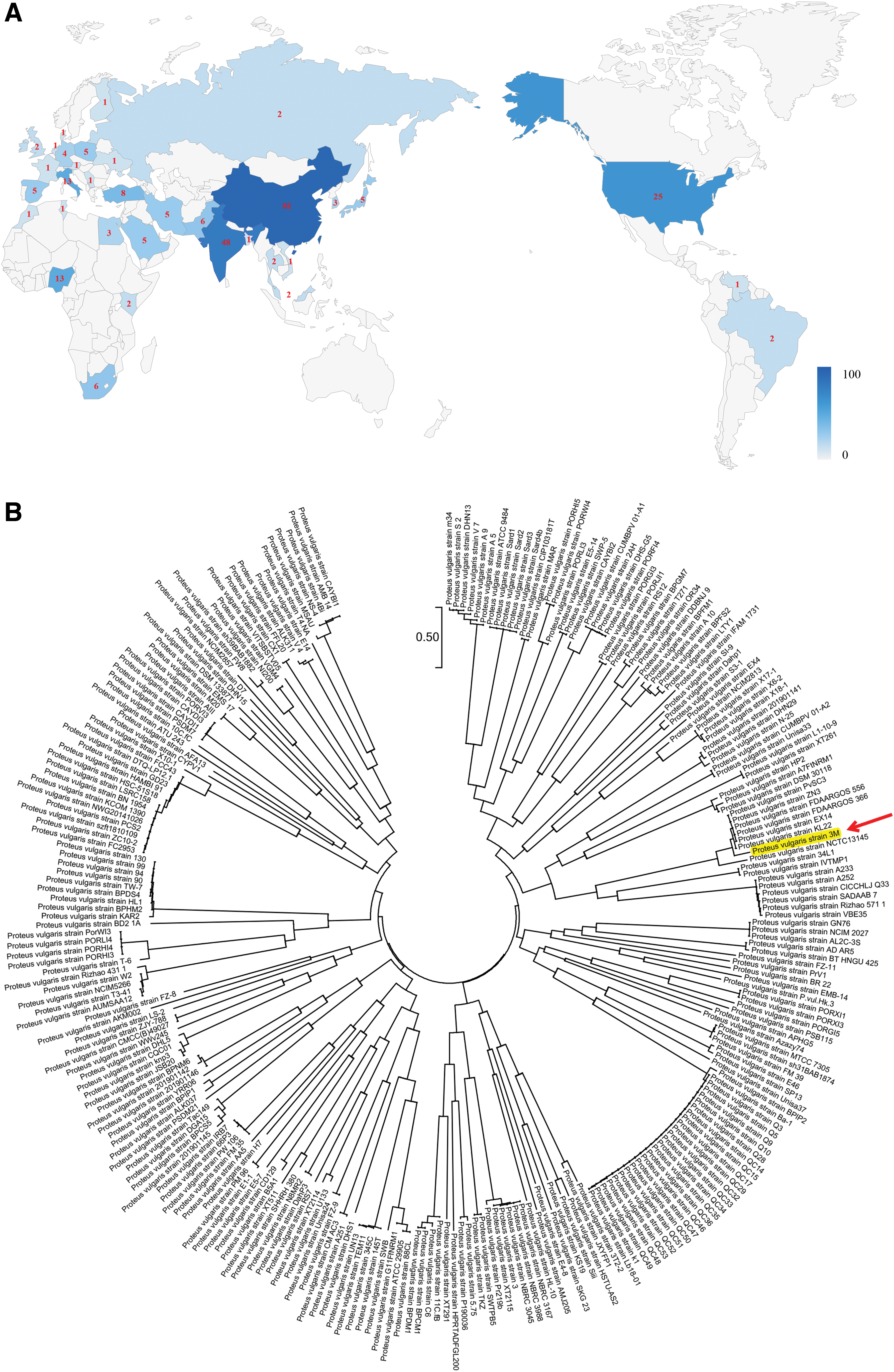
The whole genomes of seven P. vulgaris strains have been sequenced to date, and the basic characteristics of these strains are extremely similar (Table 4). PvSC3, FDAARGOS_556, and Biosolid 26 share similar genome sizes, and are all ∼4.1 Mb, whereas the genomes of P3M, NCTC13145, and FDAARGOS_366 are all ∼3.9 Mb and contain ∼3,600 genes, including 3,500 CDSs and 100 RNAs. In addition, based on the existing reports, we found that a total of 259 P. vulgaris strains have been isolated and identified and almost all the sources of P. vulgaris isolates are food or clinical origins, throughout the world (Fig. 3A and Supplementary Table S2). At present, the largest number of P. vulgaris isolates has been detected in Asia, whereas no information on P. vulgaris in Oceania has yet been published. Phylogenetic analysis further revealed the genetic relationships among the 259 P. vulgaris strains available in the GenBank database (Fig. 3B). Of note, P3M isolated from Pe. vannamei in this study was most closely related to strain KL22 isolated from sea sediment, indicating that bacteria isolated from similar habitats have closer evolutionary relationships.
General Information on All Sequenced Proteus vulgaris Strains
Two-component systems mediating multidrug resistance in P3M
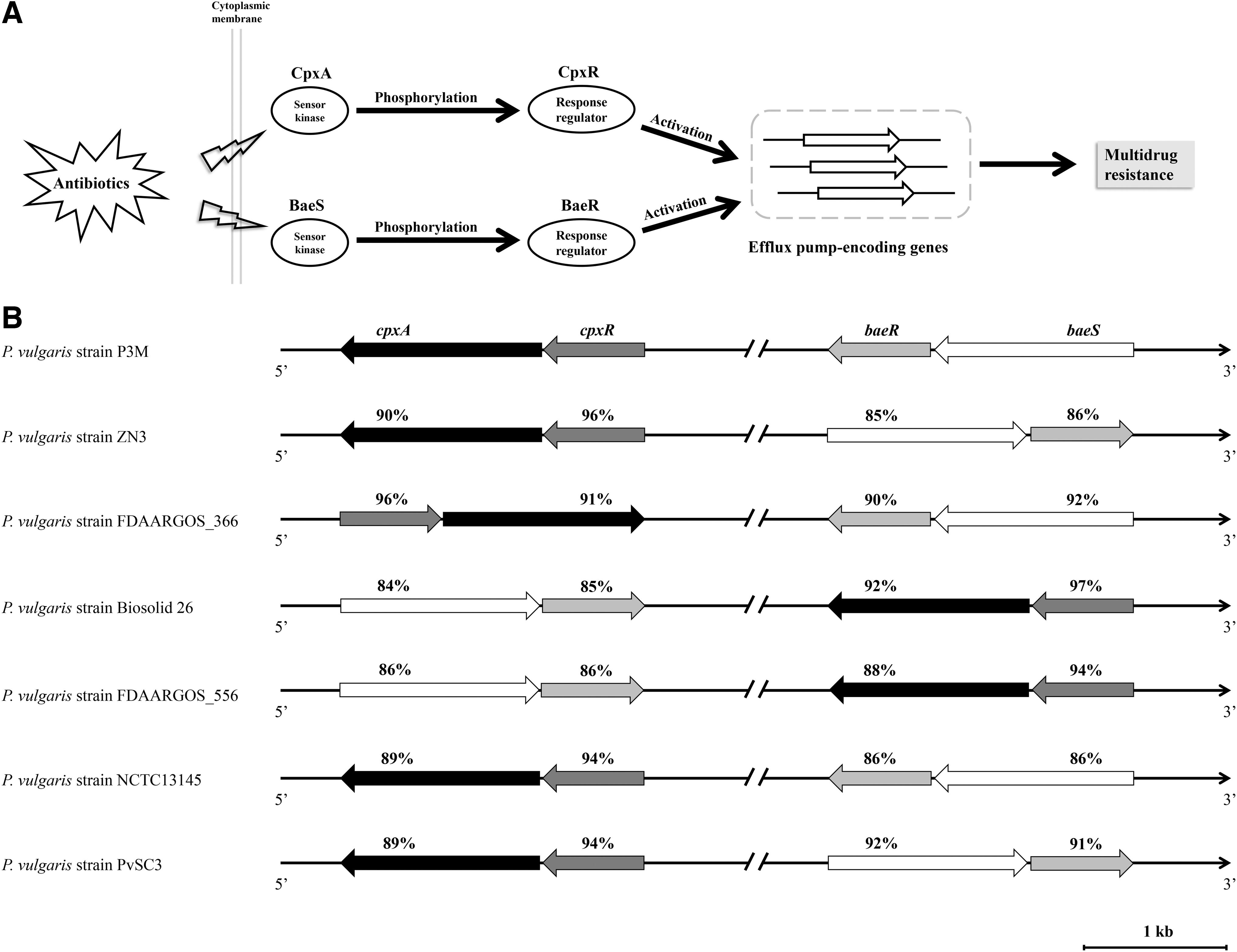
Two-component systems (TCSs), comprising a sensor kinase and a cognate response regulator, have been shown to play important roles in regulating the antibiotic resistance of various bacteria because they can be used to sense many stressful environments.53–55 In this study, CpxA–CpxR and BaeS–BaeR were identified in P3M by WGS and the prediction of ARG. As many studies have shown, CpxA and BaeS, which act as sensor kinases, rapidly respond to antibiotics outside the cell and subsequently phosphorylate CpxR and BaeR, respectively. Phosphorylated CpxR and BaeR directly activate the efficient expression of the corresponding efflux-pump-encoding genes, and the cognate proteins excrete the intracellular antibiotic and reduce the drug concentration, ultimately leading to drug resistance (Fig. 4A).56–58 These two TCSs were also detected in the other six P. vulgaris strains whose whole genomes have been sequenced and shared high sequence identity although their genetic loci in the genomes were not identical (Fig. 4B). These findings suggest that CpxA–CpxR and BaeS–BaeR are intrinsic components that may be involved in the regulation of antibiotic resistance in the species P. vulgaris. The specific regulatory roles of these two TCSs in P3M remain to be clarified.
Two-component systems mediating multidrug resistance in P3M.
Identification of possible MGEs in P3M
MGE-mediated horizontal gene transfer plays an important role in the transmission of ARGs.22,23 In our previous studies, we reported two plasmids derived from P3M, one of which carries the quinolone-resistance gene qnrD, and the other of which has specific regulatory roles. Both plasmids can be transferred into E. coli through transformation, facilitating the spread of quinolone resistance. 50 Integrons are one of the important MGEs in antibiotic-resistant cassettes, and can integrate into plasmids or chromosomes through site-specific recombination. 59 In this study, we identified six site-specific recombinases encoded in the genome of P3M (Table 5), implying that site-specific recombination and the integration of ARGs may occur in P3M. The homologous recombination of DNA mediated by direct repeats is a common genetic and evolutionary mechanism,27,60 showing the potential that antibiotic determinants could be transferred through this specific mechanism in P3M. In this study, 245 direct repeats were identified in the genome of P3M (Supplementary Table S3), among which the longest sequence involved was 5,564 bp, whereas the shortest was 110 bp. We also found that some direct repeats in the genome occur only once, whereas others occur up to dozens of times. These findings support the possibility of the transmission and integration of ARGs between different loci of the P3M genome through different MGEs.
Site-Specific Recombinases in the Genome of P3M
Discussion
P. vulgaris is one of the main species of the genus Proteus and a common opportunistic pathogen, detected both environmentally and clinically. The extensive use of antibiotics has achieved good therapeutic effects in a short time, but has also triggered the emergence of MDR bacteria, becoming the biggest challenge in the field of global public health.14,17,18 In this study, a P. vulgaris strain designated P3M was isolated from the intestinal tract of Pe. vannamei in Tianjin, China. WGS showed that P3M shares many similarities with the other six sequenced P. vulgaris strains (Table 4), whereas it differs from them insofar as 218 ARGs were identified in the P3M genome, among which 58 ARGs are closely related to the metabolic pathways of human diseases. These results imply that P3M is likely to be an MDR bacterium and the antimicrobial susceptibility results confirm this conjecture (Table 2). The determination of MICs displayed the broad antimicrobial-resistance spectrum of P3M, showing significant resistance to 16 of 28 antibiotics tested. Of note, P3M is markedly resistant to polymyxin B and colistin E, which are the therapeutic drugs of last resort for the clinical treatment of Gram-negative MDR bacterial infections.21,61,62 These results indicate that P3M is a significant clinical pathogen for its strong antibiotic resistance and pose serious threats to public health. Without doubt, this bacterium warrants further research for a more reasonable usage of antibiotics in clinical. Checkerboard analyses showed that the combination of CIP plus TET resulted in synergistic activity and reductions in the MIC of both antibiotics (Table 3). We believe that this finding will guide the future clinical treatment of diseases caused by P. vulgaris, and reduce the generation rate of MDR P. vulgaris strains to a large extent.
The worldwide occurrence of P. vulgaris strains reflects their strong propagation characteristics (Fig. 3 and Supplementary Table S2). In view of the evolutionary relationships among all isolated P. vulgaris strains and the close link between their isolation sources and human activities, it is reasonable to infer that the P. vulgaris strains isolated worldwide could spread through food chains or other viable channels, making the problem of antibiotic resistance more serious.63,64 Many inherent functional elements (TCSs, MGEs, etc.) in the genome of species P. vulgaris enable the bacteria to better survive and to withstand the adverse effects of environmental antimicrobial drugs, leading to the increase of drug resistance and the possibility of horizontal transfer of ARGs.
In conclusion, we report for the first time an important foodborne MDR polymyxin-resistant P. vulgaris isolate, P3M. The worldwide spread and proliferation of species P. vulgaris further exacerbate the clinical problem of multidrug resistance. Continued research into the regulatory mechanisms of TCSs, the basic biology of MGEs, and the horizontal transfer mechanism of ARGs is required to understand the MDR characteristics of P. vulgaris in depth, and to provide reference data for the clinical treatment of Gram-negative MDR bacteria and the rational use of antibiotics in the future.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by the Key Projects of the National Natural Science Foundation of China (Grant No. 41831287) and the Tianjin Postgraduate Scientific Research Innovation Project (Grant No. 2019YJSB042).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.