
Abstract
Antimicrobial resistance (AMR) plays an important role in the pathogenesis and spread of Clostridioides difficile infection (CDI). Many antimicrobials, such as fluoroquinolones, have been associated with outbreaks of CDI globally. This study characterized AMR among clinical C. difficile strains in Thailand, where antimicrobial use remains inadequately regulated. Stool samples were screened for tcdB and positives were cultured. C. difficile isolates were characterized by toxin profiling and PCR ribotyping. Antimicrobial susceptibility testing was performed by agar incorporation, and whole-genome sequencing and AMR genotyping were performed on a subset of strains. There were 321 C. difficile strains isolated from 326 stool samples. The most common toxigenic ribotype (RT) was RT 017 (18%), followed by RTs 014 (12%) and 020 (7%). Resistance to clindamycin, erythromycin, moxifloxacin, and rifaximin was common, especially among RT 017 strains. AMR genotyping revealed a strong correlation between resistance genotype and phenotype for moxifloxacin and rifaximin. The presence of erm-class genes was associated with high-level clindamycin and erythromycin resistance. Point substitutions in the penicillin-binding proteins were not sufficient to confer meropenem resistance, but a Y721S substitution in PBP3 was associated with a 4.37-fold increase in meropenem minimal inhibitory concentration. No resistance to metronidazole, vancomycin, or fidaxomicin was observed.
Introduction
C
C. difficile can be separated into different ribotypes (RTs) by amplifying the intergenic spacer region between the 16S and 23S rRNA genes. 7 This method has been used widely due to its simplicity and high discriminating power. 8 Important C. difficile RTs include C. difficile RT 027, an A+B+CDT+ strain associated with outbreaks of severe CDI in North America and Europe in the early 2000s, 9 C. difficile RT 078, another A+B+CDT+ strain, was associated with the zoonotic transmission, 10 and C. difficile RT 017, a tcdA-negative (A−B+CDT−) strain, was associated with global outbreaks since 1995. 6
Although resistance to the antimicrobials used for the treatment of CDI (metronidazole, vancomycin, and fidaxomicin) is rare, 11 resistance to other antimicrobials plays an important role in the pathogenesis and spread of CDI. While intrinsic resistance to cephalosporins was probably responsible for an increase in the rate of CDI worldwide in the 1980s, 12 resistance to clindamycin, new-generation fluoroquinolones, rifamycins, and tetracyclines has been associated with CDI outbreaks. 13 These antimicrobials are also associated with an increased risk of developing CDI in general. 14 Strict regulation of antimicrobials is a successful measure to control CDI. In the United States, such regulation has lead to a significant decrease in CDI cases and CDI-related deaths over the last decade. 15 Fluoroquinolone regulation in Australia has resulted in a relatively low prevalence of fluoroquinolone-resistant organisms, 16 including C. difficile. 17
Several studies have reported an association between antimicrobial resistance (AMR) genotypes and phenotypes for various antimicrobials. The most common clindamycin resistance determinant is the erm(B) gene, which methylates and protects 23S rRNA from the antimicrobial. 13 However, concordance between the presence of the erm(B) gene and the resistance phenotype is low. 18 A subsequent study suggested that erm(B) may only be associated with high-level clindamycin resistance and thus the mechanism underlying low-level clindamycin resistance remains unknown. 19 Carbapenem resistance is also poorly described. So far, only imipenem resistance has been characterized and is associated with point mutations on the penicillin-binding proteins PBP1 and PBP3. 20 On the contrary, fluoroquinolone and rifaximin resistance are well characterized and are associated with point substitutions in the quinolone resistance determining region on the DNA gyrase subunits (GyrA and GyrB) and RNA polymerase subunit B (RpoB), respectively. 21
In previous studies, the epidemiology of CDI in Thailand has been characterized by a high prevalence of A−B+CDT− and an absence of A+B+CDT+ strains, as well as a high prevalence of NTCD, which may play a protective role against the development of CDI.22–24 C. difficile strains isolated in Thailand, especially C. difficile RT 017, had a high prevalence of resistance to many antimicrobial groups, similar to other pathogenic bacteria in the country25,26 reflecting possibly poor antimicrobial stewardship in the country. 27 This study provides an update on the characterization and antimicrobial susceptibility of C. difficile isolated from a tertiary hospital in Bangkok, Thailand.
Materials and Methods
Isolation and characterization of C. difficile
This study was undertaken on 326 diarrheal stool samples collected from patients with a high index of suspicion of CDI at Siriraj Hospital, a large teaching hospital in Bangkok, Thailand, during 2017–2018. All stools were first positive for tcdB using the BD Max Cdiff assay (Becton Dickinson), as a part of routine investigations at Siriraj Hospital, and these were sent to a reference laboratory in Perth, Western Australia, for further investigation.
At the reference laboratory, stools were processed as previously described. 28 Briefly, a portion of each stool sample was directly inoculated on ChromID C. difficile agar (bioMérieux, Marcy I'Etoile, France) and incubated anaerobically for 48 hours before the putative C. difficile colonies were identified. The remainder of each sample underwent enrichment culture in supplemented brain/heart infusion broth, followed by ethanol shock to increase the sensitivity of the culture process. C. difficile isolates were characterized by PCR ribotyping, performed as described by Stubbs et al., with a QIAxcel Advanced System capillary gel electrophoresis platform (QIAGEN, Venlo, The Netherlands). 7 The banding patterns were compared with a local database consisting of 80 internationally recognized RTs, including 15 reference RTs from the European Centre for Disease Prevention and Control. This method can differentiate similar RTs, such as RTs 014 and 020 (Supplementary Fig. S1). Patterns that did not match strains in the database were given an internal nomenclature. Detection of tcdA and tcdB, and the binary toxin genes, was performed as described by Kato et al. 29 and Stubbs et al., 5 respectively. All NTCD isolates in this study were confirmed as such by PCR as described by Braun et al. (lok PCR). 2
All stool samples were tested also for colonization with multiple C. difficile strains. Briefly, DNA extraction was performed on all enrichment broths. DNA was then screened with either tcdB 29 or lok 2 PCR based on the toxin profile of the first C. difficile strain isolated from the specimen. For example, a specimen previously positive for TCD was screened with lok PCR for NTCD and vice versa. All PCR-positive broths were recultured and up to 30 putative C. difficile colonies per broth were selected and characterized by toxin gene profiling. An isolate with a different toxin profile from the first strain was treated as the second strain from the same sample and underwent further characterization by PCR ribotyping.
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing (AST) was performed by agar incorporation, as described by the Clinical and Laboratory Standards Institute, against the eight antimicrobials listed in Supplementary Table S1. 30 C. difficile ATCC 700057, Bacteroides fragilis ATCC 25285, Eubacterium lentum ATCC 43055, and B. thetaiotaomicron ATCC 29741 were included as controls. Susceptibility results were interpreted using the minimal inhibitory concentration (MIC) breakpoints listed in Supplementary Table S1.30–34 C. difficile strains resistant to at least three antimicrobial classes were classified as multidrug resistant (MDR). Resistance to clindamycin and erythromycin was considered resistance to a single class (macrolide/lincosamide/streptogramin B [MLSB]).
Whole-genome sequencing, high-resolution typing, and antimicrobial resistance characterization
A subset of 37 C. difficile strains was selected for whole-genome sequencing. Genomic DNA was extracted, sequenced on an Illumina HiSeq platform that generated 150 bp pair-end reads with a median coverage of 73 × , and characterized by multilocus sequence typing (MLST) as previously described. 35 Clade assignment of a new sequence type (ST) was confirmed by comparing the average nucleotide identity (ANI) with C. difficile strains 630 (clade 1, accession AM180355) and R20291 (clade 2, accession FN545816) using FastANI. 36 Accessory AMR genes were identified by interrogating the read files with SRST2 version 0.2.0 against the ARG-ANNOT database version 3.37,38 Draft annotated genomes were interrogated on Artemis version 17.0.1, and additional accessory genes identified. 39 Known point substitutions associated with resistance to carbapenems (substitution in penicillin-binding proteins PBP1 and PBP3), fluoroquinolones (substitution in the GyrA and GyrB subunits of the gyrase enzyme), and rifaximin (substitution in the RpoB enzyme)20,21 were also identified using SRST2 as previously described. 19
Data availability
All sequence data were submitted to the European Nucleotide Archive under BioProject PRJEB40974, accessions ERS5247348–ERS5247384 (Supplementary Tables S2 and S3). Two newly characterized resistance determinants were submitted to the Nomenclature Center for MLSB Genes, 40 and the sequences were submitted to GenBank (accessions MW269959 [erm(52) gene] and MW269960 [mef(H) gene]). Genomes containing the prototypes of these genes were submitted to GenBank under BioProject PRJNA679085, accessions JADPMU000000000 [MAR225, carrying erm(52)] and JADPMT000000000 [MAR272, carrying mef(H)].
Statistical analyses
All statistical analyses were performed using online tools by Social Science Statistics available at https://www.socscistatistics.com/ A p-value ≤0.05 was considered statistically significant.
Results
Characterization of Thai C. difficile
A total of 296 C. difficile strains were initially isolated from the stools and another 25 strains were identified from the cocolonization screening process, yielding a total of 321 C. difficile strains. Of these, 221 (68.85%) were positive for tcdA and tcdB (A+B+CDT−), 58 (18.07%) were positive for tcdB only and had a deletion in tcdA (A−B+CDT−), 3 (0.93%) were positive for all toxin genes (A+B+CDT+), and 39 strains (12.15%) were negative for all toxin genes (A−B−CDT−, NTCD). A list of samples with multiple C. difficile strains is provided in Supplementary Table S4.
The 321 C. difficile strains belonged to 63 RTs, 19 of which were internationally recognized. The remaining RTs were given an internal nomenclature (prefix “QX-“ or “KI-“). The prevalence of the common RTs is summarized in Table 1. The most common TCD strain was C. difficile RT 017 (A−B+CDT−), followed by RTs 014 and 020 (both A+B+CDT−). The most common NTCD was C. difficile RT 010.
Ribotypes of Three Hundred Twenty-One Clostridioides difficile Strains from Thailand, by Toxin Profile
RTs designated with “QX” and “KI” were RTs that did not match the internationally recognized RTs in the database and were given internal nomenclature.
RT, ribotype.
Characterization of a novel binary toxin-positive C. difficile strain
One C. difficile strain was positive for all three toxin genes (A+B+CDT+) and had a unique ribotyping pattern. According to the MLST scheme, this isolate was characterized as the novel ST 692 within evolutionary clade 1. However, pairwise ANI analysis showed that this strain was more closely related to C. difficile R20291 (clade 2, ANI = 99.17%) than C. difficile 630 (clade 1, ANI = 98.89%).
Antimicrobial susceptibility of Thai C. difficile
AST results are shown in Table 2 and the MIC distribution of selected six antimicrobial classes is displayed in Fig. 1. Based on the MIC value, clindamycin-resistant C. difficile strains could be divided into two groups: those with MIC ≥32 mg/L (n = 97) and those with MIC <32 mg/L (n = 166). There was a strong correlation between high-level clindamycin resistance and erythromycin resistance: 95 strains (97.94%) that had clindamycin MIC ≥32 mg/L were also resistant to erythromycin, while only 16 strains (9.64%) in the other group were resistant to erythromycin (Cohen's κ = 0.857).
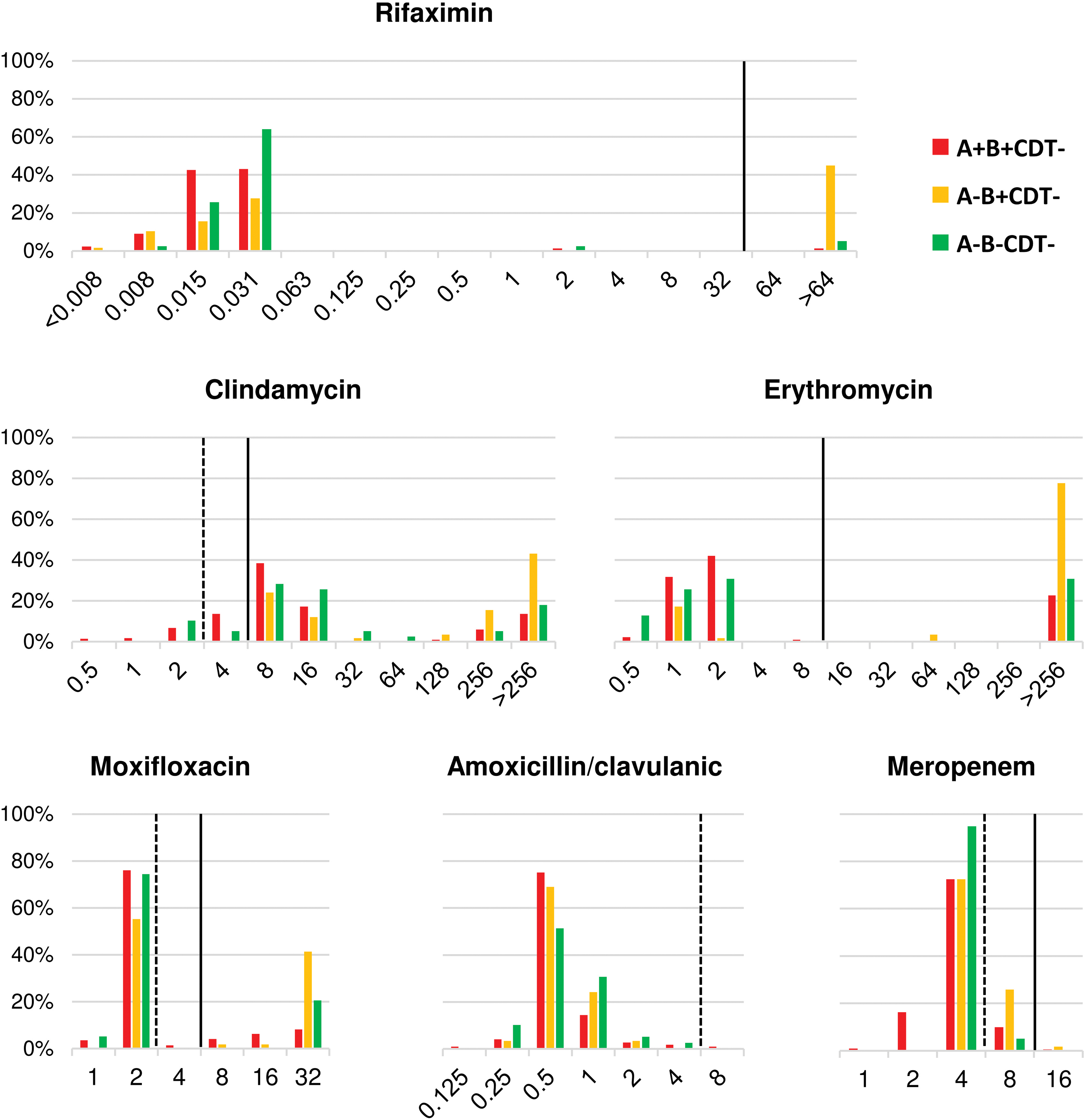
Minimal inhibitory concentration distribution for six antimicrobials against 321 strains of Clostridioides difficile in Thailand. C. difficile strains were classified according to their toxin gene profiles: A+B+CDT−, red; A−B+CDT−, yellow; A−B−CDT−, green. The number of A+B+CDT+ C. difficile was low (n = 3) and was excluded. Breakpoints for intermediate resistance (I) and resistance (R) are shown by broken and solid lines, respectively. Color images are available online.
Antimicrobial Susceptibility of Three Hundred Twenty-One C. difficile Strains from Thailand
MIC, minimal inhibitory concentration.
When classified by toxin gene profiles, resistance to clindamycin, erythromycin, moxifloxacin, and rifaximin was more prevalent among A−B+CDT− C. difficile, all belonging to RT 017, than A+B+CDT− and NTCD (Fig. 1). Twenty-nine (9.03%) C. difficile strains were MDR, 26 (8.10%) of which were C. difficile RT 017. The remaining strains were NTCD (n = 2) and A+B+CDT− C. difficile (n = 1). All MDR strains were resistant to MLSB (both clindamycin and erythromycin), moxifloxacin, and rifaximin. One MDR strain was also resistant to meropenem (RT 017, MIC = 16 mg/L).
AMR genotypes in Thai C. difficile
A summary of MIC values and AMR genotypes of 37 sequenced C. difficile strains is available in Supplementary Table S3. Thirty-one C. difficile strains had high-level resistance to clindamycin: 23 strains carried erm(B), 5 carried erm(G), and 3 carried a gene encoding an rRNA adenine N(6)-methyltransferase protein. This gene was given the name erm(52). Of the 23 erm(B)-positive strains, 19 carried the gene on transposon Tn6194 (82.61%), while the other 4 (17.39%) carried the gene on Tn6189. No erm-class genes were identified among strains with low-level clindamycin resistance. The concordance between the presence of erm-class genes and high-level clindamycin resistance was 100%. A gene encoding a macrolide efflux protein was identified in two strains with high-level erythromycin resistance (MIC >256 mg/L) and only low-level clindamycin resistance, and given the name mef(H). No significant genotypic resistance determinants were identified in strains with low-level clindamycin resistance.
Twenty-five sequenced strains were resistant to moxifloxacin (MIC 8–32 mg/L). Of these, the T82I substitution in GyrA and the D426V substitution in GyrB were found in 23 strains and 1 strain, respectively. No known point substitutions were found in one strain with low-level moxifloxacin resistance (MIC 8 mg/L), as well as in all moxifloxacin-susceptible strains (97.37% concordance). There were H502N and R505K substitutions in RpoB in all 23 rifaximin-resistant strains and none in the susceptible strains (100% concordance).
Twelve strains had an A555T substitution in PBP1 and another seven had a Y721S substitution in PBP3. A multiple linear regression analysis suggested that the Y721S substitution in PBP3 was associated with a 4.37-fold increase in meropenem MIC (95% confidence interval: 2.78–5.96, adjusted R2 = 0.516, t = 5.521, p < 0.0001), while the A555T substitution in PBP1 was not associated with the change in meropenem MIC (t = −1.127, p = 0.268).
Discussion
This study provides an update on the molecular epidemiology and antimicrobial susceptibility of C. difficile strains circulating in Thailand. It also explores the genomic basis of important AMR in these strains. The overall epidemiology of C. difficile was similar to the previous studies.22–24 The majority of A+B+CDT− strains belonged to C. difficile RTs 014 and 020, all A−B+CDT− strains belonged to C. difficile RT 017, and most NTCD belonged to C. difficile RTs 009, 010, and 039. Three binary toxin-positive strains were found in this study, one of which was C. difficile RT 078. The epidemic C. difficile RT 027 remained absent in Thailand despite its successful spread in some other regions. 41
Why C. difficile RT 027 has failed to spread and to establish in Thailand remains unknown. One possible reason is that the successful spread of this RT was mainly due to its resistance to fluoroquinolones, which provided a selective advantage over other less resistant RTs. 42 Although there is high consumption of fluoroquinolones, such as levofloxacin, in the country, 43 Thailand already harbors C. difficile RT 017, another epidemic RT, many of which are resistant to fluoroquinolones as well as other antimicrobials. 13 Thus, it may have been difficult for C. difficile RT 027 to compete with this local RT compared with other regions.
Although C. difficile RT 027 was not identified, a possible relative of this hypervirulent strain, ST 692, was isolated. The MLST result was unusual, as it was classified into clade 1 despite carrying a complete CDT locus, a common feature in C. difficile clades 2 and 5 but rare in clade 1. 44 Thus, an ANI analysis was performed. In a previous study, C. difficile strains from the same clade generally shared >99% ANI. 44 Thus, the ANI results suggested that this newly characterized strain was a member of clade 2 rather than clade 1, as expected from the toxin gene profile. The average ANI between clades 1 and 2 in a previous study was around 98%, which further supports the results. 44 Clades 1 and 2 C. difficile are closely related and share a large proportion of housekeeping gene alleles used in the MLST scheme. As a result, it may be difficult to properly discriminate these two clades by MLST. The use of ANI analysis, which involves the whole genome rather than a specific set of housekeeping genes, can help in the correct classification of some borderline strains, as shown in a previous study. 44 According to the ANI analysis, it is more likely that C. difficile ST 692 belongs to clade 2 and is related to C. difficile RT 027.
A discordance between culture results and the result of a conventional real-time tcdB PCR was observed in 44 stool samples. The false-positive rate of the real-time PCR method (13.50%) was comparable with the previous report comparing tcdB PCR with a similar culture method but without the colonization screening step, 28 suggesting that the additional screening step does not increase the yield of the culture method, although it may help identify stool samples with multiple C. difficile strains. This false-positive rate also highlights the importance of patient clinical data or additional tests to improve the accuracy of CDI diagnosis. In the latest guidelines for the treatment and diagnosis of CDI, tcdB PCR in combination with another diagnostic test is recommended, commonly a toxin antigen detection kit, for a proper diagnosis of CDI and the use of stand-alone tcdB PCR should be interpreted with caution. 45
AMR in C. difficile mainly impacts the pathogenesis of CDI. To cause the disease, C. difficile must tolerate the presence of antimicrobials in the intestinal lumen while the microbiota perishes. 13 Many successful C. difficile lineages have been characterized with increased resistance to at least one major drug group. 13 In this study, C. difficile RT 017, the most prevalent RT, had greater resistance to MLSB (both clindamycin and erythromycin), moxifloxacin, and rifaximin than other RTs. It was also the most common MDR C. difficile strain. C. difficile RT 017 has been reported also to be the most prevalent RT with significant resistance to many antimicrobials in other parts of Thailand. 24 This particular RT has been associated with resistance to at least six antimicrobial groups, 13 which may account for its successful global spread. 6 As regulation of antimicrobial use has reduced the impact of C. difficile in many countries,15,17 a similar approach should be effective in Thailand.
All erm(B)-positive C. difficile strains carried the gene on two well-characterized erm(B)-positive transposons: Tn6189 and Tn6194, the latter being found also in C. difficile M68, a C. difficile RT 017 strain widely used as a reference in genomic studies. 13 Tn6194, the most prevalent transposon in this study, is capable of interspecies transfer, most notably between C. difficile and Enterococcus faecalis. 46 This emphasizes another aspect of AMR in C. difficile; its possible role as a reservoir of AMR genes for other pathogenic bacteria residing in the colon.
Previously, the low concordance between the presence of the erm(B) gene and an MLSB resistance phenotype was reported, 18 likely due to the presence of multiple resistance mechanisms. However, another study suggested that the erm(B) gene may be associated only with high-level MLSB resistance. 47 We also observed separation between C. difficile strains with high-level and low-level clindamycin and erythromycin resistance (Fig. 1). Upon genomic analysis, there was a strong correlation between the presence of an erm-class gene [erm(B), erm(G), and erm(52) genes] and high-level clindamycin resistance, which is usually accompanied by high-level erythromycin resistance, supporting the earlier study. 47 Resistance determinants were not identified among strains with low-level clindamycin resistance, however, this underestimation is likely irrelevant, as the median clindamycin MIC in this population (8 mg/L) remained lower than the clindamycin level in stools (∼240 mg/g of stool). 48 Besides MLSB, a separation between strains resistant and susceptible to rifaximin and fluoroquinolones was observed (Fig. 1). The concordance between resistant phenotype and known genotype was high, similar to a previous study. 18
Compared with the study at the same hospital in 2015, there was no difference in the overall resistance prevalence, 31 however, there was a slight increase in meropenem MICs and the emergence of carbapenem resistance. Carbapenem resistance in C. difficile is poorly characterized, possibly due to its rare occurrence. A previous study reported an association between point substitutions in PBP1 and PBP3 and high-level imipenem resistance, although these substitutions do not confer meropenem resistance. 20 We confirmed that neither the substitution on PBP1 nor PBP3 was associated with meropenem resistance. However, linear regression analysis suggested that the Y721S substitution in PBP3 may have contributed to a 4.3-fold increase in meropenem MIC. Thus, this substitution could be a part of a multistep meropenem resistance mechanism. Indeed, two C. difficile strains in this study had meropenem MICs of 16 mg/L (resistance breakpoint ≥16 mg/L), one of which was confirmed to have the Y721S substitution in PBP3.
C. difficile remained susceptible to metronidazole, vancomycin, and fidaxomicin, similar to the other parts of the world. 49 Thus, these antimicrobials should remain effective treatments for CDI. There was a slight increase in vancomycin MIC reaching the clinical breakpoint, consistent with a previous study, 31 however, this should have little impact on the treatment of CDI as the stool vancomycin concentration remains far greater than the MIC (>2,000 mg/L vs. 2 mg/L). 50 The increase in vancomycin MIC in this study is in contrast to other hospitals in Thailand and this could reflect usage of vancomycin at the study site. 24 Overuse of vancomycin can lead to the emergence of vancomycin-resistant Enterococcus spp., which can have a devastating effect on patients.51,52 Therefore, vancomycin usage should be carefully monitored.
Conclusion
A−B+CDT− C. difficile and NTCD remained prevalent in Thailand. Few binary toxin-positive strains (A+B+CDT+) were identified; one belonging to a known epidemic lineage and the other a novel strain related to C. difficile RT 027. The most common strain was C. difficile RT 017 (A−B+CDT−), a large proportion of which was resistant to MLSB, moxifloxacin, and rifaximin. Many strains were also MDR. Such resistance may have played a role in the success of C. difficile RT 017 in Thailand. There was a strong concordance between the presence of erm-class genes and high-level clindamycin resistance, as well as a significant concordance between point substitutions in gyrase subunits and RpoB with fluoroquinolone and rifaximin resistance, respectively. Resistance to antimicrobials suitable for the treatment of CDI was not detected.
Ethical Conduct of Research Statement
This study was approved by the Human Research Ethics Committee of The University of Western Australia (reference file RA/4/20/4704) and the Siriraj Institutional Review Board (protocol number 061/2558 [EC1]).
Footnotes
Acknowledgment
Parts of this study were performed using the facilities provided by the Pawsey Supercomputing Centre (Perth, Western Australia).
Disclosure Statement
T.V.R. has received grants from Cepheid, Merck, Otsuka, Roche, Sanofi, and Summit for work outside that in this report. Other authors have no conflicts of interest to declare.
Funding Information
This work was supported by Mahidol University (Mahidol Scholarship to K.I.) and the National Health and Medical Research Council of Australia (Peter Doherty Biomedical Early Career Fellowship [APP1138257] to D.R.K).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.