
Abstract
The global emergence of antibiotic resistance, especially in Gram-negative bacteria, is an urgent threat to public health. Inevitably, considering its extensive use and misuse, resistance toward ciprofloxacin has increased in almost all clinically relevant bacteria. This study aimed to investigate the transcriptome changes at a high concentration of ciprofloxacin in Escherichia coli. In brief, 1,418 differentially expressed genes (DEGs) were identified, from which 773 genes were upregulated by ciprofloxacin, whereas 651 genes were downregulated. Enriched biological pathways reflected the upregulation of biological processes such as DNA damage and repair system, toxin/antitoxin systems, formaldehyde detoxification system. With kyoto encyclopedia of genes and genomes pathway analysis, higher expressed DEGs were associated with “LPS biosynthesis,” “streptomycin biosynthesis,” and “polyketide sugar unit biosynthesis.” Lower expressed DEGs were associated with “biosynthesis of amino acids” and “flagellar assembly” pathways. After treatment of ciprofloxacin, lipopolysaccharide (LPS) release was increased by two times, and the gene expression level of LPS synthesis was elevated (p < 0.05) in both reference and clinical strains. Our results demonstrated that transient exposure to high-dose ciprofloxacin is a double-edged sword. Cautions should be taken when administering high-dose antibiotic treatment for infectious diseases.
Introduction
Ciprofloxacin is among the most commonly prescribed class of fluoroquinolones, which is widely used in the therapy of mild-to-moderate urinary and respiratory tract infections caused by Enterobacteriaceae. However, growing resistance to these agents leads to the decline of their clinical efficacy. Before the evolution of antibiotic resistance, bacteria frequently undergo response and tend to develop a state of adaption to the antibiotic.
Antibiotic exposure has been shown to promote the emergence of resistant bacteria. 1 There are many environments or human tissues during therapy where bacteria are exposed to low concentrations of antibiotics (less than minimum inhibitory concentration [MIC]). Many studies have shown the profound transcriptomic changes in bacterial cells and their potential contributions to the evolution of resistance when exposed to low levels of ciprofloxacin.2–4
In the therapeutic use of ciprofloxacin, levels of ciprofloxacin in tissues or serum can be reached or exceed the MIC. Long-term exposure to ciprofloxacin above the MIC will kill the susceptible bacteria and select resistant mutation. However, little is known about the influence of a transient high-level (>MIC) exposure of ciprofloxacin on transcription in bacteria. This study investigated the transcriptome changes under the action of high concentration ciprofloxacin in Escherichia coli.
Materials and Methods
Bacterial strain and growth conditions
Twenty nonduplicate clinical isolates of E. coli from patients were selected randomly in tertiary hospitals of Harbin Medical University. Clinical isolates and E. coli K12 strain MG1655 were grown in 250 mL flasks with Luria-Bertani (LB) medium in a shaking incubator at 37°C and 250 rpm overnight. For ciprofloxacin treatment, overnight cultures were subcultured to early exponential phase and treated with 2 μg/mL ciprofloxacin (Sigma) for 20 min. Bacterial abundances are expressed as colony-forming units (CFU) per volume of fluid medium. The number of CFU is multiplied by the dilution factor and expressed in CFU/mL.
This study involved only bacterial strains that were already isolated and thus, no ethics approval was necessary.
Antimicrobial susceptibility testing
MICs of ciprofloxacin were determined using the broth microdilution method according to the Clinical and Laboratory Standard Institute (CLSI) guidelines. 5 The MIC for each clinical isolate was measured at least in triplicate and the median of all results was reported.
RNA isolation, cDNA library construction, and Illumina sequencing
Total RNA was extracted from E. coli cells with Qiagen RNA protect bacterial reagent (Qiagen, Hilden, Germany) and purified with Qiagen RNeasy Plus Mini Kit (Qiagen) following the manufacturer's instruction. After assessment of RNA purity and integrity, RNA samples were treated with Epicentre Ribo-zero rRNA Removal Kit (Epicentre, Madison, WI), followed by RNA fragmentation, synthesis of the first and second strands of DNA, and appendices of sequencing adapter. Then the selected fragments by Agencourt AMPure XP Beads (Illumina, Inc., San Diego, CA) were amplified, followed by library purification, enrichment, and quality determination.
Accordingly, six cDNA libraries (three from ciprofloxacin-treated E. coli and three from non-ciprofloxacin-treated E. coli) were constructed and subjected to Illumina Hiseq 2500 sequencing platform. The genome accession number was NC_000913.3. Clean reads were obtained by filtering out low-quality adaptor-polluted and high content of unknown base read from raw reads. The clean reads were aligned to the assembled reference to NCBI Nr database (nonredundant protein) by Bowtie 2 with a mismatch of 2. 6 Searches were conducted by the BLASTx 7 program with an e-value cutoff of 1e−5.
Differentially expressed gene identification, gene ontology and kyoto encyclopedia of genes and genomes pathway enrichment, and statistical analysis
The DEGseq R package, 8 DEseq, 9 and edgeR 10 software were used for the identification of differentially expressed genes (DEGs) induced by ciprofloxacin by pairwise comparison. Gene expression level was measured in fragments per kilobase of exon per million reads mapped (FPKM). The threshold values of significant DEGs were set at |logFC| ≥ 1, FDR (false discovery rate) ≤0.05, and adjusted p-value ≤0.05. Hierarchical clustering analysis of DEGs was performed with log10(FPKM +1). The gene ontology (GO) knowledgebase is a source of information on the functions of genes. Kyoto encyclopedia of genes and genomes (KEGG) is a database resource for understanding high-level functions and utilities of the biological system from large-scale molecular datasets. GO term enrichment and KEGG pathway analysis of DEGs was performed. GO and KEGG pathway terms with adjusting p-value <0.05 were considered significant.
Quantitative reverse transcription PCR
To detect the effect of ciprofloxacin on the transcription of selected genes associated with motility, 1,000-fold diluted overnight cultures of E. coli MG1655 were cultured to early exponential phase. Ciprofloxacin (2 μg/mL) was added to the bacterial culture and incubated at room temperature for 30 min. Bacterial pellets were harvested by centrifugation and resuspended in phosphate-buffered saline containing 100 mg/L lysozymes (Sigma-Aldrich) and incubated for 10 min at room temperature.
Total RNA was extracted with TRIzol reagent (Invitrogen), digested with Turbo DNase-free kit (Ambion, Austin, TX) to remove contaminating DNA, and quantified with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE). cDNA was synthesized with 100 ng of total RNA by using the M-MLV reverse transcriptase (Promega, Madison, WI) and random hexamers (Takara).
Expression of genes associated with lipopolysaccharide (LPS) biosynthesis (Supplementary Table S1) was quantified using a two-step quantitative reverse transcription PCR (qRT-PCR) analysis. qRT-PCR was carried out with SYBR (TaKaRa), in a 20 μL volume using a CFX96 qRT-PCR detection system (Bio-Rad, Hercules, CA) under the following conditions: 50°C for 2 min, initial denaturation at 95°C for 10 min, and 45 cycles of 15 sec at 95°C and 1 min at 60°C.
Results were analyzed with SDS software (v. 2.2 Applied Biosystems). The data were normalized to the endogenous reference gene (mdh) and analyzed by the threshold cycle method (2−ΔCT). The fold change of genes was calculated by 2−ΔΔCT. This experiment was repeated at least three times with independently isolated RNA samples. The primers used in qRT-PCR are shown in Table 1.
Primers Used in this Study
Statistical analysis
The statistical difference in gene expression of qRT-PCR results was analyzed by Student t test and FDR was also calculated to correct the p-value. It was considered to be statistically significant when p-value <0.05 or q-value <0.05.
Quantitation of LPS
Endotoxin contents in the supernatant of bacteria with or without ciprofloxacin treatment were detected by limulus amebocyte lysate (LAL) assay kit (Thermo Scientific) according to the manufacturer's instruction. In brief, bacteria were microcentrifuged. The supernatant was harvested and added in triplicate to equal volume of LAL in a pyrogen-free microtiter plate. The mixture was incubated at 37°C for 10 min, and 100 μL of chromogenic substrate solution was added and color development was terminated by the addition of 20% acetic acid. The optical density was measured at 410 nm.
Results
Bacteria growth
The growth of MG1655 before and after ciprofloxacin treatment is shown in Fig. 1. Bacterial strains were grown from a cell concentration of 7.53 Log10CFU/mL to 8.00 Log10CFU/mL within 60 min under normal growth conditions. After treatment with 2 μg/mL ciprofloxacin, bacterial strains cell concentration of 3.66 Log10CFU/mL at 30 min and 3.24 Log10CFU/mL at 60 min.

Survival curves of MG1655 before and after ciprofloxacin treatment.
Overview of sequencing data
The quality of sequencing data from ciprofloxacin treated MG1655 and the controls are summarized and shown in Table 2. A high mapping rate (>70%) was detected after BLASTx alignment to reference data in NCBI (Table 3). Also, almost all of the reads were mapped to coding DNA sequence genome regions (>98%; Supplementary Fig. S1). With the Pearson correlation analysis, we showed there were high repeatabilities and minor variations between samples (Supplementary Fig. S2).
Summary of Sequencing Data Quality
Q20, Q30: percentage of bases with Phred value >20, 30, respectively.
Summary of Clean Reads Mapped to Reference Genome
Read-1 and -2: the forward and reverse sequence of pair-end sequencing; “+” and “−”: alignment to the + strand and − strand of the reference genome.
Transcript annotation
With the annotation to the Nr database, we identified transcripts including DEGs, sRNA, and novel transcripts. Among these transcripts, a total of 647 low expressed and 797 high expressed differentially expressed transcripts (DETs) were identified (Supplementary Fig. S3A). With transformed log10(FPKM+1) values, hierarchical clustering analysis of all DETs was performed and 6 distinct clusters were got (Supplementary Fig. S3B). Among those 797 high expressed transcripts, 6 sRNAs, 18 novel transcripts, and 773 DEGs were included (including lexA, recA, recX, tisB, gyrA, gyrB, stpA, and dnaN), and the low expressed transcripts consisted of 2 novel transcripts and 645 DEGs (including thrA-C, flgA-B, and fliF-L; Supplementary Table S1).
GO and KEGG pathway enrichment analysis
To map the functional annotation and pathways related to these DEGs, we performed the GO and KEGG pathway enrichment analysis of DEGs induced by ciprofloxacin treatment.
Results showed high expressed DEGs encoding “putative fimbrial-like adhesin protein” (e.g., ybgD, yraH, yehA, yfcV, and csgAB operon) were enriched in the pilus, cell part, cell adhesion, and so on. Higher expressed DEGs encoding the 50S/30S ribosomal subunit protein (e.g., frmRAB, S10, and spc ribosomal protein operons) and DEGs encoding regulator protein that represses frmRAB operon, which encodes a formaldehyde detoxification system. DNA recombination and repair protein (recA) were associated with GO terms related to ribosome complex, cellular amide metabolic process, and so on.
Toxic membrane persister formation peptide gene (tisB) was also higher expressed, which is regulated by lexA and participates in toxin/antitoxin systems. Some DEGs were associated with DNA repair or replication pathways, for example, dnaN and holD (Supplementary Table S2 and Fig. 2A). Lower expressed DEGs encoding proteins related to flagellum development (e.g., flgG, flgB, and fliL operons), including GO terms of the cilium or flagellum-dependent cell motility, bacterial-type flagellum, and bacterial-type flagellum basal body (Supplementary Table S2 and Fig. 2B).
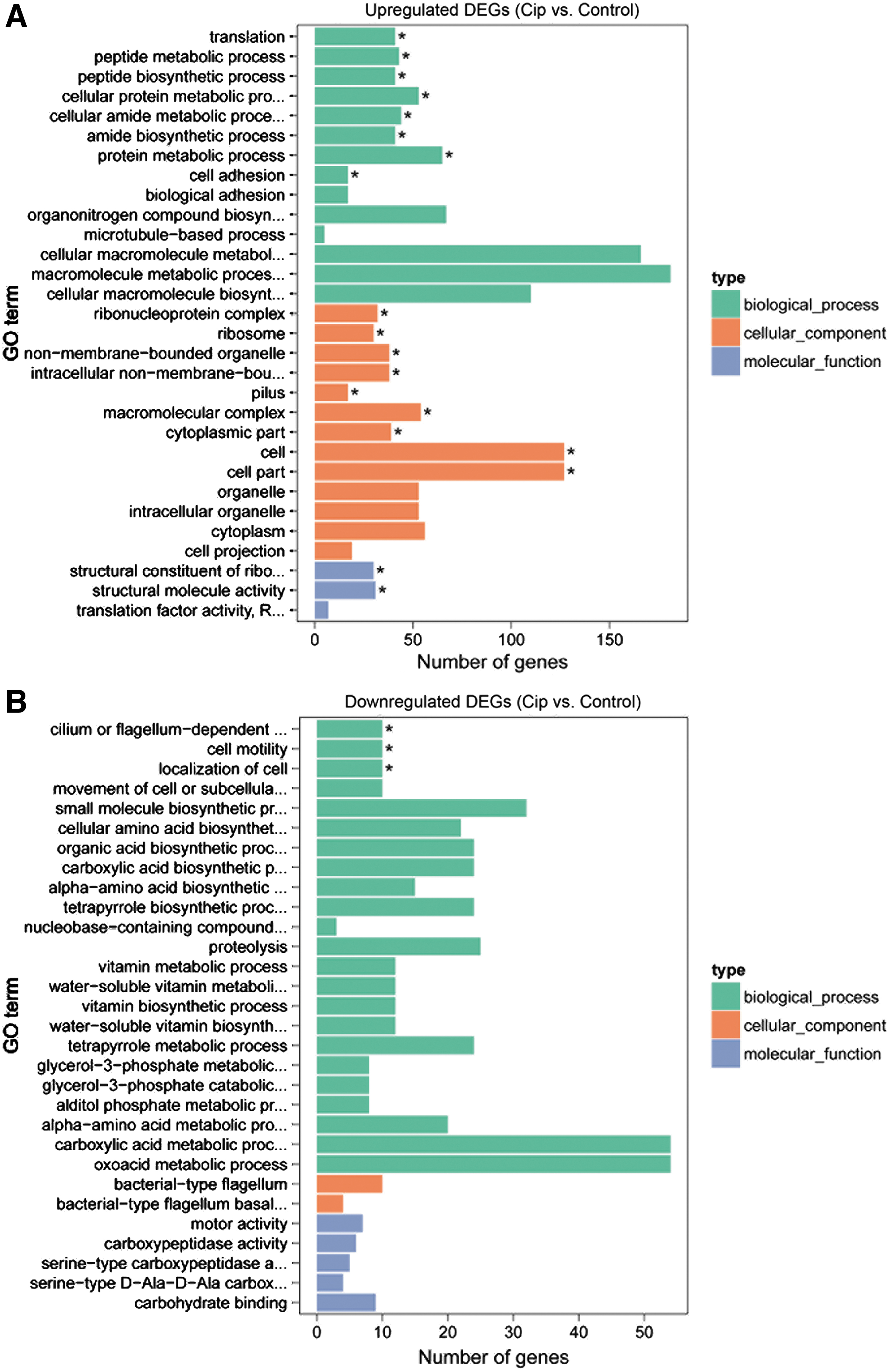
The top 30 enriched GO terms of DEGs in Escherichia coli induced by ciprofloxacin.
With KEGG pathway analysis, higher expressed DEGs and lower expressed DEGs were associated with two significant pathways, respectively (adjusted p-value <0.05; Supplementary Table S3 and Fig. 3). However, these terms were too few to understand the mechanisms related to ciprofloxacin responses in MG1655. So we selected the top 20 KEGG pathways to analyze related mechanisms independently. Accordingly, we found higher expressed DEGs (e.g., S10, spc, recR, ruvABC, waa, rfbABC, and frmRAB operon) were enriched into the biosynthesis of secondary metabolites, including pathways of the ribosome, methane metabolism, thiamine metabolism, and so on (Supplementary Table S3 and Fig. 3A).
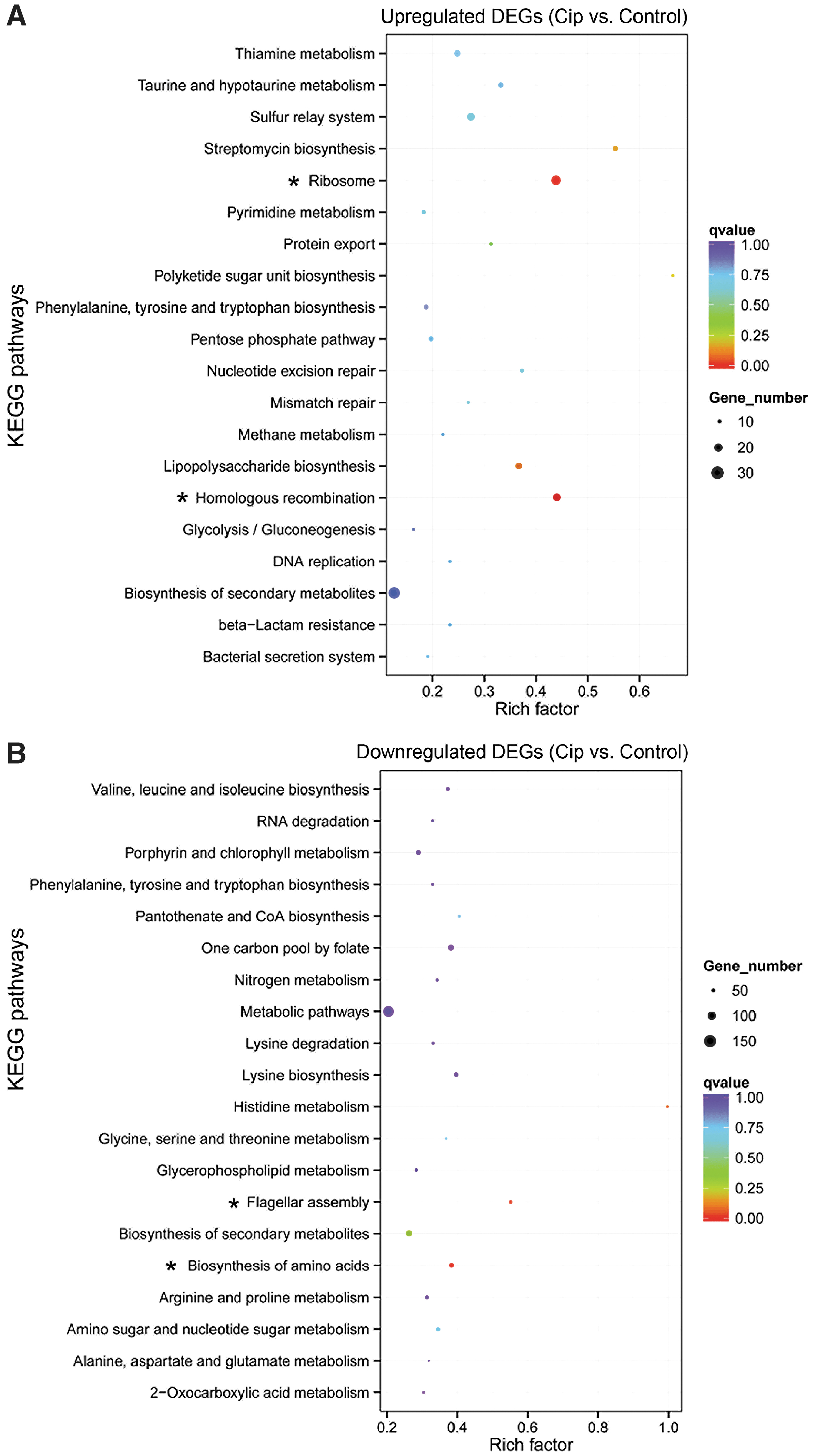
The top 20 enriched KEGG pathways of DEGs in E. coli induced by ciprofloxacin.
waa operon (includes 10 genes) was associated with LPS biosynthesis. rfbABC operon (encodes 3 polypeptides rfbA, rfbB, and rfbC) was related to streptomycin biosynthesis and polyketide sugar unit biosynthesis. Most of the lower expressed DEGs (e.g., thrABC, flgG, flgB, and fliL operons) were enriched into pathways related to amino acids metabolism and flagellum development, including pathways of biosynthesis of amino acids, flagellar assembly, arginine and proline metabolism, and so on. The nar operon (encodes 4 polypeptides NarG, NarH, NarJ, and NarI), and GltBD operons (encodes GltB and GltD) were related to nitrogen metabolism (Supplementary Table S3 and Fig. 3B).
Real-time PCR verification
Among the selected clinical strains of E. coli, 15 were susceptible and 5 were resistant to ciprofloxacin. The MIC value of ciprofloxacin is shown in Table 4. The expression level of recA was elevated from 9.13 ± 2.20 to 62.25 ± 8.46 after ciprofloxacin treatment in MG1655 (p < 0.001). The mRNA expression level of LPS synthesis genes, waaB, waaP, and waaG were also elevated after ciprofloxacin treatment in MG1655. After 20 min of treatment with ciprofloxacin, the level of waaB increased from 0.76 ± 0.24 to 2.24 ± 0.60 (p < 0.05), waaP increased from 0.81 ± 0.12 to 2.34 ± 0.46 (p < 0.01) and waaG increased from 0.91 ± 0.18 to 2.16 ± 0.45 (p < 0.05) (Fig. 4).

The expression levels of recA and LPS synthesis genes after high concentration ciprofloxacin treatment. After 20 min of treatment with ciprofloxacin, the levels of recA, waaB, waaP, and waaG (*p < 0.05, **p < 0.01, ***p < 0.001). LPS, lipopolysaccharide.
Minimum Inhibitory Concentration of Ciprofloxacin in Clinical Isolates of Escherichia coli
MIC, minimum inhibitory concentration.
After ciprofloxacin treatment, the endotoxin-related genes of 15 clinical sensitive strains of E. coli were changed, and the endotoxin-related genes of four strains (4/15) were highly expressed (Fig. 5A), and the average expression level of endotoxin-related genes increased in 15 clinical sensitive strains of E. coli (Fig. 5B). LPS synthesis genes were increased in both clinical sensitive strains and MG1665 after ciprofloxacin treated. Among the five clinical-resistant strains of E. coli, the concentration of 2 μg/mL ciprofloxacin was lower than the minimum MIC of clinical-resistant strains, so it failed to significantly change the expression level of the endotoxin-related genes in clinical drug-resistant strains of E. coli (Fig. 5A).
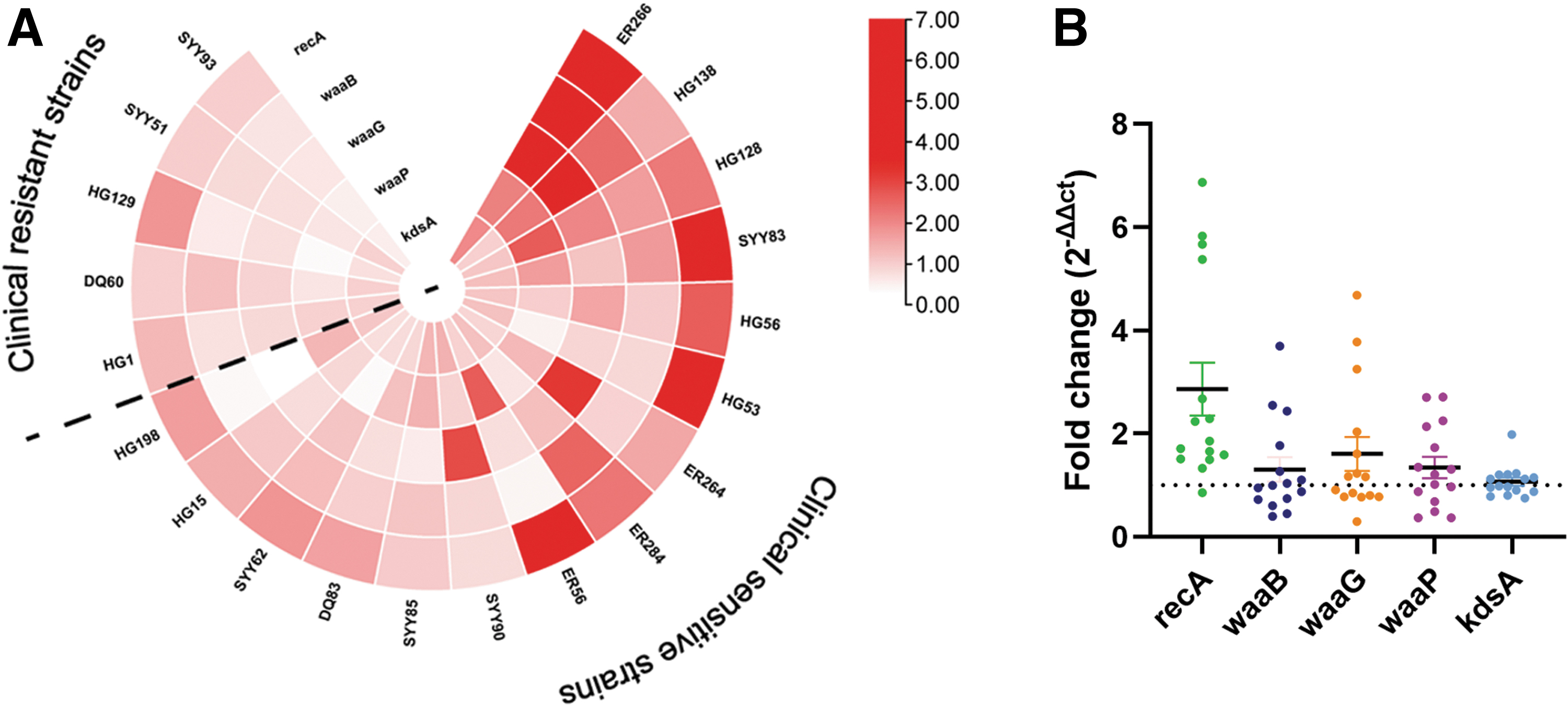
The fold change in the expression of endotoxin-related genes in clinical strains of E. coli after treatment with 2 μg/mL ciprofloxacin.
Determination of LPS release after ciprofloxacin treatment
Through the analysis of the GO and KEGG database, we found that LPS biosynthesis and waa operon expression were high. So the release of LPS in MG1655 supernatant was determined after 20 min ciprofloxacin treatment. It is shown that bacterial LPS release was increased after ciprofloxacin treatment (p < 0.01) (Fig. 6A). The LPS release of clinical sensitive strains was also increased after 2 μg/mL ciprofloxacin treatment as MG1655 (Fig. 6B).

Endotoxin level released by E. coli after high concentration ciprofloxacin treatment for 20 min.
Discussion
The increase of ciprofloxacin resistance may lead to a high treatment failure rate of bacterial infections and an increase in infection rates of antibiotic-resistant bacteria.11,12 The canonical mechanisms of ciprofloxacin resistance include genetic mutations of gyrA and parC or horizontal dissemination of antibiotic resistance genes, which protects or modifies the targets of fluoroquinolone. 13
Many studies have shown that transcriptional response in bacterial cells induced by a subinhibitory concentration of antibiotics contributes to the formation of bacterial resistance.14,15 However, little is known about the influence of a transient high-level exposure of ciprofloxacin on bacterial transcriptome. High-dose antibiotic therapy has been a common treatment for infection. The concentration of 2 μg/mL is approximate to the maximum concentration in tissue when treating infections. 16 For E. coli MG1655 used in this study, the concentration is higher than the MIC of ciprofloxacin.
High concentration of antibiotics caused cellular damage by DNA damage and decrease of motive force
Ciprofloxacin induces DNA damage such as breaking double-stranded DNA and forking stalled replication, which triggers genetic exchanges. 17 In this study, we identified some genes related to DNA damage, including gyrA, gyrB, stpA, ygbT, parE, and dnaN, which were elevated in ciprofloxacin-treated cells. Among these genes, stpA encodes an H-NS-like protein, which constrains DNA supercoils and influences DNA topology. 18 gyrA gene contributes to DNA relaxation during DNA replication and mediates the breakage and reunion of DNA strand. 19 High expression of these genes indicated that high dose of ciprofloxacin induced DNA damage in E. coli cells.
Besides, high concentrations of antibiotics can inhibit bacterial motility. In ciprofloxacin-treated E. coli, gene clusters associated with curli or flagellum biogenesis and metabolisms were lower expressed (Supplementary Table S2). Under particularly harmful conditions, decreased expression of exercise genes is regarded as a universal means of self-protection through energy conservation.20,21
The thrABC operon or threonine operon consists of four threonine biosynthesis genes, which are mediated by the level of threonine and isoleucine in cytoplasm, which is essential for cell growth.22,23 The fliL operon consists of seven adjacent genes encode fliL, fliM, fliN, fliO, fliP, fliQ, and fliR, which is required for flagellar biogenesis and normal cell division. 24 The downregulation of thrABC and fliL operons in ciprofloxacin-treated E. coli indicated that ciprofloxacin treatment disrupted normal development.
SOS system, toxin/antitoxin system, and formaldehyde detoxification system activation, and their significance in drug resistance
The bacterial SOS response is a global response to DNA damage in which the cell cycle is arrested and DNA repair and mutagenesis are induced. Many studies have demonstrated that subinhibitory concentrations of quinolones activate the SOS pathway by DNA damage and induce ciprofloxacin resistance.25,26 SOS pathway includes the transcription of stress response genes, such as SOS regulon (recA and lexA), and the genes encode DNA repair proteins and polymerases.27,28 Our current analysis identified the upregulation of SOS-response genes, including recA gene and its regulator recX, lexA.
Dörr et al. demonstrated that SOS response-induced tisB “toxin” DNA damage-inducible toxin, controlling the production of multidrug tolerant cells, 27 which can also decrease bacterial growth and cause multidrug resistance.29,30 In this study, lexA regulated genes tisB, ybfE, and ydjM were higher expressed after ciprofloxacin treatment. Toxin/antitoxin systems were also higher expressed upon ciprofloxacin exposure. High-dose ciprofloxacin induced a rapid increase of the tisB toxin within 20 min during exponential phase, indicating that bacterial are capable of responding to the fluoroquinolone stress promptly and tend to form persister cells before completing one cycle of binary fission.
The frmRAB operon and rfb clusters were higher expressed after ciprofloxacin treatment by RNA-seq analysis. The frmRAB operon encodes a formaldehyde detoxification system in E. coli and is responsible for glutathione (GSH)-mediated formaldehyde detoxification,31,32 which prevents deleterious effects of formaldehyde accumulation in E. coli. 33 We speculate that cellular GSH induced by ciprofloxacin treatment triggered the formaldehyde degradation pathway and consequently increased the expression of frmRAB operon in E. coli. RfbABC operon is essential for the synthesis of deoxysugar and O-antigen, and multicellular development. 34
These demonstrated that ciprofloxacin-treated E. coli cells produced a cellular protection system, including enhanced formaldehyde detoxification and auto-repair mechanisms. These were in accordance with the activated SOS response and the ciprofloxacin tolerance or resistance in E. coli cells. In our study, the DEGs in rfbABC operon were also enriched into “streptomycin biosynthesis” pathway (Supplementary Table S3).
Significance of increased LPS synthesis induced by a high concentration of antibiotics
Many studies have reported that subinhibitory concentration of quinolones can increase the LPS released by the bactericidal lysis of bacteria.35–37 RNA-seq analysis in this study revealed that genes related to biosynthesis of LPS were higher expressed, such as rfb gene cluster, waa operon, and kdsA. Further qRT-PCR and LPS assay confirmed that both the LPS release of bacteria and expression of LPS synthesis-related genes increased under the action of ciprofloxacin at bactericidal concentration. For clinical strains, after treatment with high concentration of ciprofloxacin, the endotoxin-related genes of all clinical sensitive strains changed, and the expression level of recA gene increased, indicating that the internal DNA of E. coli was damaged and the SOS system was activated, suggesting that the change of endotoxin may depend on the activation of SOS system.
These results suggest that high-dose fluoroquinolone not only induces bacterial canonical stress response enabling bacterial populations to survive a high concentration of antibiotics but also increases LPS production at the genetic level by increasing gene expression. LPS is a major constituent of the outer membrane. Overproduction of LPS stimulates monocytes and macrophages to produce large amounts of proinflammatory mediators that cause sepsis. 38 It is known that LPS can trigger inflammatory responses against pathogens in the human body, even at very low doses (1 ng/kg body mass). 39
Therefore, it is important to reduce the toxicity of bacterial endotoxin while using ciprofloxacin for antimicrobial treatment. In addition to the application of endotoxin scavengers such as polymyxin B to neutralize the endotoxin produced, it is important for agents designed to inhibit the expression of endotoxin genes are the potential to promote therapeutic efficiency and reduce the risk of endotoxemia.
Conclusions
We confirmed by RNA sequencing that a high dose of ciprofloxacin to the exponential phase of E. coli induces DNA damage, decreases biosynthesis of amino acids and flagella assembly within 20 min exposure. In addition to the bacterial damaging effects, a high concentration of ciprofloxacin can upregulate the transcriptional level of the SOS system and toxin–antitoxin system, which are involved in the formation of persistent and resistant bacteria. While helping bacteria acquire adaptive resistance, a high dose of ciprofloxacin may also produce bacteria with a high transcriptional expression of LPS, which should be vigilant during the antibiotic therapy. These results demonstrated that transient exposure to high-dose ciprofloxacin is double-edged. Cautions should be taken when administering high-dose antibiotic treatment for infectious disease.
Footnotes
Acknowledgment
The authors thank Wenjing Li for valuable technical advice.
Authors' Contributions
Y.M.F., F.M.Z., and X.Q.Z. designed the study. Experiments were performed by R.S., P.H., Q.T.M., Q.Z., and W.L.Z. Data analysis were performed by X.Y.L. and R.S. Y.M.F., R.S., Q.T.M., P.H., and X.Q.Z. wrote the article. All authors read and approved the final article.
Availability of Data and Materials
We promise that all the data supporting the set of our conclusions are included in this article and its additional files. The complete RNA sequencing data generated and analyzed in this study have been submitted to the Genome Expression Omnibus (GEO) database, and the accession number is GSE152445 (![]() ).
).
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the National Science and Technology Major Project from Ministry of Science and Technology of the People's Republic of China (MOST; 2017ZX10201301-003-005) and National Natural Science Foundation of China (NSFC; 31370164 and 81701613). The sponsors of this study had no involvement in the conduct of research or decisions regarding this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.