
Abstract
Mycobacterium spp. intimidated mankind since time immemorial. The triumph over this organism was anticipated with the introduction of potent antimicrobials in the mid-20th century. However, the emergence of drug resistance in mycobacteria, Mycobacterium tuberculosis, in particular, caused great concern for the treatment. With the enemy growing stronger, there is an immediate need to equip the therapeutic arsenal with novel and potent chemotherapeutic agents. The task seems intricating as our understanding of the dynamic nature of the mycobacteria requires intense experimentation and research. Targeting the mycobacterial cell envelope appears promising, but its versatility allows it to escape the lethal effect of the molecules acting on it. The unique ability of hiding (inactivity during latency) also assists the bacterium to survive in a drug-rich environment. The drug delivery systems also require upgradation to allow better bioavailability and tolerance in patients. Although the resistance to the novel drugs is inevitable, our commitment to the research in this area will ensure the discovery of effective weapons against this formidable opponent.
Introduction
The discovery of the Mycobacterium tuberculosis (MTB) by Robert Koch in 1982 was a major conquest in the history of modern medicine. In 2020, an estimated 1.3 million patients (uncertainty interval: 1.2–1.4 million HIV-negative individuals) died of tuberculosis (TB). 1 Before the emergence of COVID-19 (coronavirus disease 2019), MTB led the list of single pathogens causing considerable mortality. 1 COVID-19 has led to an increase in the vulnerability to TB probably due to weakening of the TB health services (TB testing, reporting, and treatment). 2
Since the 1950s, a wide array of extremely potent and effective therapeutic drugs have been used for the treatment of TB and other mycobacterial diseases. Despite being highly efficient against the mycobacterial cell, all of these drugs have eventually been subjected to resistance posed by mycobacterial strains. Thus, the goal for the perfect treatment of TB remains unaccomplished. Considering the alarming rise of multidrug-resistant TB (MDR-TB), extensively drug-resistant TB (XDR-TB), and the recently described totally drug-resistant TB (TDR-TB), the World Health Organization (WHO) has mentioned MDR-TB as a public health security threat. 3
A challenging aspect in the development of new drugs against Mycobacterium spp. is the presence of slow or nongrowing strains called persisters, which are not only resistant to host immune response but also to bactericidal antibiotics. Moreover, considerable proportions of the antimycobacterial drugs are only available as parenteral formulations and also have serious adverse effects, which result in reduced patient compliance. Therefore, there is a continuing need for new antimycobacterial drugs that are available orally, have fewer side effects, and can deal with the bacteria during the latency.
With the exhaustive research going on for the past two decades for new drugs and their targets, the mycobacterial cell envelope has re-emerged as a crucial target. Some of these drugs have already been approved, while many others are in the pipeline. Drugs such as delamanid and pretomanid have also been found to be active even against the dormant strains or persisters. 4
However, the development of drugs acting on the cell envelope is full of challenges and limitations. Genotypic and phenotypic variations in the clinical MTB strains modulate their susceptibility to the promising drug candidates along with their inherent properties, their pharmacokinetic–pharmacodynamic properties, or their interactions with the existing and the upcoming antimycobacterial and antiretroviral drugs.5,6
In this regard, the chemical nature, mechanism of action, and spectrum of activity of many novel molecules acting on the mycobacterial cell envelope have been extensively discussed in the literature. However, the impediments in their path of becoming the preferred drug for treatment have not been addressed comprehensively. The present review specifically focuses on the challenges for the therapeutic options targeting mycobacterial cell envelope and a brief account of the progress in the development of these promising agents.
The Complex Organization of Mycobacterial Cell Envelope
The cell wall and cell membrane play an essential role in the growth, virulence, and survival of bacteria in the body of the host. 7 MTB is a unique bacterium, not only in its pathogenesis and disease spectrum but also due to its peculiar cell wall structure (Fig. 1), which makes it different from other bacteria. 8 The major part of the cell wall consists of lipids with few carbohydrates and proteins, making it an effective barrier against most of the hydrophilic compounds, including some antimicrobial agents.
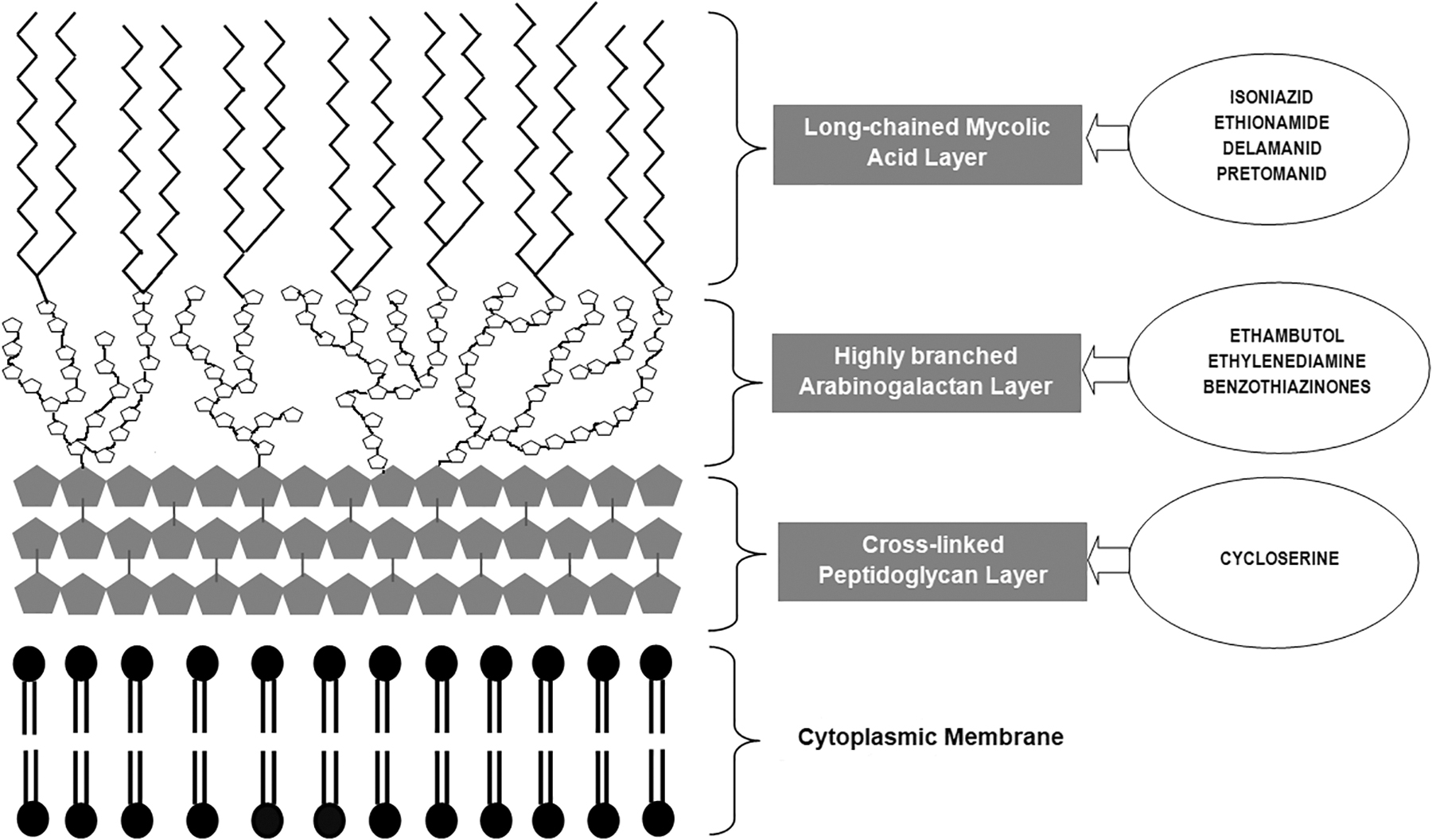
A schematic diagram of mycobacterial cell envelope depicting the mycolyl–arabinogalactan–peptidoglycan complex and cell membrane along with the drugs targeting those structures.
The unique structure of the cell envelope of MTB contains a cytoplasmic membrane (also referred to as the inner membrane) over which lies the mycolyl–arabinogalactan–peptidoglycan complex, which functions as the skeleton of the cell wall. This complex consists of three major structural layers (from inside outward), viz. (1) peptidoglycan layer (present in the form of a cross-linked network); (2) an arabinogalactan-based polysaccharide layer (present in a highly branched form); and (3) the long-chain mycolic acid layer, which is the hallmark of a mycobacterial cell wall. 9
Outside this, there is a lipid bilayer called the outer membrane. This comprises free glycolipids and inert waxes that intercalate the mycolic acid layer. The outermost component is the capsule consisting of proteins and polysaccharides, which serve as the first point of contact of the mycobacterial cell with the surrounding environment. 8
The organization of the mycobacterial cell envelope is complex and asymmetric in nature, which largely contributes to the general drug-resistance phenotype of the bacterium.10,11
Current Status of Drug Development Against Mycobacterial Cell Envelope
The cell wall and membrane when intact is an integral barrier for survival and virulence in both active and static MTB strains. Therefore, extensive research is ongoing to study various components of the cell envelope that can act as potential therapeutic targets. 12 These chemotherapeutic agents disrupt the normal architecture of the envelope, by either targeting one or more of its structural components or the enzymes involved in their synthesis, thus turning an otherwise impermeable cell wall into a breached, permeable one.8,13 The mechanisms of action of various U.S. Food and Drug Administration (FDA)-approved antimicrobials acting on the mycobacterial cell envelope have been summarized in Table 1.
The List of Cell Wall-Acting Agents Approved for Treatment of Mycobacterial Infections
ACP, enoyl-acyl carrier protein; AcpM, acyl carrier protein; Alr, alanine racemase; KasA, β-ketoacyl-ACP synthase.
The most important landmark in the history of TB had been the miraculous breakthrough of effective antitubercular chemotherapeutic agents. Among the first discovered antibiotics against MTB were streptomycin and para-aminosalicylic acid (PAS) in the year 1944, while the earliest discovered cell wall inhibitor drug was isonicotinic acid hydrazide (isoniazid [INH]) by Domagk in the year 1952. In 1960, ethambutol (EMB) was isolated, and it replaced PAS used in the treatment of TB, as it not only led to a reduction in overall treatment duration but also it had fewer side effects and thus was better tolerated. 14 A brief history of the discovery of anti-TB drugs and their incorporation into the treatment regimen has been depicted in Fig. 2.
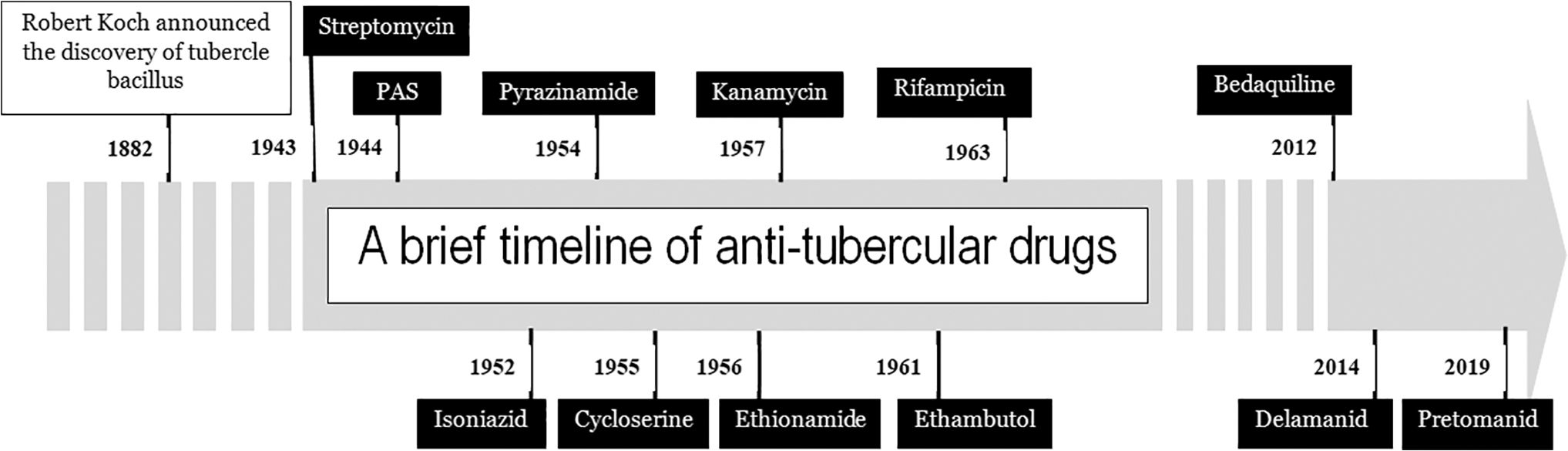
A brief timeline of therapeutic drugs introduced in the treatment of tuberculosis (names of the drugs that target the mycobacterial cell wall are mentioned at the bottom of the arrow). PAS, para-aminosalicylic acid.
Cell wall-acting agents already approved for therapeutic use
Isoniazid
Since its discovery, INH had been the backbone of anti-TB chemotherapy and had always been used as the first-line drug. It is available in the oral formulation and has good oral bioavailability, and accumulates in deep tissues and organs in high concentrations. 13 One of the limitations of INH is that it exerts bactericidal activity only against actively replicating MTB strains, and it does not have any activity against dormant, stationary, or latent strains. 15 INH occurs as a prodrug, which, after entering the MTB cells, gets activated by the enzyme mycobacterial catalase-peroxidase, encoded by the katG gene.
As a result of this activation, INH-derived intermediates and other reactive oxygen species are generated, which in turn combine with NADH to inhibit InhA (an NADH-dependent enoyl-acyl carrier protein [ACP] reductase involved in mycolic acid biosynthesis). 16 The other intracellular target for INH is a complex of an acyl carrier protein (AcpM) and a β-ketoacyl-ACP synthase (KasA), an enzyme that is also involved in the synthesis of mycolic acids. 17 Resistance to INH has been a worrying issue across the globe.
The aggregated drug-resistance data from 156 countries have revealed that on average, 11.4% (95% confidence interval [CI] 9.4–13.4) previously treated patients and 7.4% (95% CI 6.5–8.4) of new cases between 2003 and 2017 had INH-resistant TB, and its overall prevalence (regardless of concomitant rifampicin resistance) varied from 0.7% (9.6–11.9) to 27.2% (24.6–29.9) based on previous treatment history and geographical locations. 18 Resistance to this drug occurs primarily because of the mutations happening in the different genes (katG, inhA, ahpC, kasA, and ndh) and also their promoter regions.19,20
Recently, a novel INH-resistance mechanism (drug inactivation) has been described in some MTB isolates with the ability to acetylate the drug. 21 Acute adverse effects of INH mainly include neurological symptoms, while chronic ones are more frequently observed, and present as hepatotoxicity and peripheral neuropathy. 22
Ethionamide
Ethionamide (ETH) is a highly potent mycobactericidal drug introduced in the early 1950s but later restricted as a second-line anti-TB drug due to some of the worrisome adverse reactions. 23 It is an analog of INH, containing thioamide group. 24 It has a similar target, that is, ACP reductase, but its mode of action on the target is different from the inhibitory actions by INH. 23 As a prodrug, it needs to get activated by a mono-oxygenase enzyme that is NADPH and O2 dependent, and uses flavin adenine dinucleotide (FAD) as a cofactor. 25 Once activated, the oxidized metabolites of ETH and NAD+ form adducts that down regulate the function of InhA enzyme, thus inhibiting the mycolic acid synthesis and disrupting the cell wall integrity. 25
Until recently, it has been hypothesized that the generation of ETH-NAD+ active adduct involves formation of an iminoyl radical from the putative sulfinic acid as an intermediary step in ETH-bioactivation. However, in the absence of evidence on the existence of sulfinic acid intermediate, the role of a novel S-oxide ETH intermediate (that behaves as a “ketene-like” compound and ultimately leads to the formation of iminoyl radical) has been proposed. 26 ETH is highly active against both drug-susceptible and drug-resistant MTB strains, but it tends to develop rapid resistance when used as monotherapy. 25
The main contributors to the resistance are an array of genes such as ethR, pknF, and mshA (with its product mycothiol).23,27 Besides, this crossresistance of INH and ETH has been reported in patients treated with INH but who never received ETH. 28 The major side effects include gastrointestinal disturbances and hepatotoxicity.23,25 A better tolerated propyl analog of ETH with similar potency was discovered and has been named as prothionamide.
Ethambutol
EMB is a first-line anti-TB drug that was discovered in 1961. Along with INH, rifampicin, and pyrazinamide, this drug is administered as an oral preparation to patients with drug-susceptible TB. It is bacteriostatic in nature, and it also acts on nontuberculous mycobacteria such as Mycobacterium avium complex and Mycobacterium kansasii.13,29 EMB exerts its action by inhibiting the enzyme arabinofuranosyl transferase, which plays an important role in arabinogalactan synthesis.
Failure to synthesize arabinogalactan, in turn, leads to inhibition of the mycolyl–arabinogalactan–peptidoglycan complex, thus disrupting the firm cell wall of mycobacteria and making it more permeable. 30 Some recent studies have revealed that EMB can also interfere with peptidoglycan biosynthesis by competitive inhibition of the glutamate racemase (MurI) enzyme. 31
The resistance to EMB is a complex phenomenon, and involves mutations in a multitude of genes such as embCAB operon (encodes the enzyme arabinofuranosyl transferase) and Rv3806 (mutation in this gene results in increased intracellular accumulation of decaprenyl phosphoryl arabinose that competes with EMB for its binding sites). 32 The major adverse effect of EMB is ocular toxicity in the form of axial or periaxial retrobulbar neuritis.33,34
Cycloserine
It is a bacteriostatic antibiotic with activity against MTB. Chemically it is a cyclic analog of D-alanine.35,36 It is an analog of alanine, and competes with and inhibits the two important enzymes responsible for core formation in the peptidoglycan layer: D-alanine:D-alanine ligase (Ddl) and alanine racemase (Alr). Alr converts L-alanine to D-alanine, whereas Ddl helps incorporating the alanine into the pentapeptide core of peptidoglycan. Thus by inhibiting Alr and Ddl, cycloserine inhibits L-alanine conversion to D-alanine and also restricts integration of alanine into the peptidoglycan layer.35,36
Cycloserine has been reclassified recently by the WHO to be used for drug-resistant TB cases (MDR-TB, XDR-TB) as a part of the second-line regimen. It does not crossreact with other first-line or second-line antimycobacterial drugs.35,36 Another advantage of cycloserine is that very few cases of drug resistance have been reported. 36 Cycloserine resistance results from mutations in several genes encoding for alanine transporter CycA (Rv1704c), the L-alanine dehydrogenase Ald (Rv2780), and the Alr. 37
Major limitations in its use include its neurotoxicity and the lack of reliable susceptibility testing methods. The adverse effects including anxiety, agitation, depression, psychosis, and, rarely, seizures may be seen even with the recommended therapeutic dose required for its antimycobacterial activity. 35
Delamanid
It is a derivative of nitro-dihydro-imidazooxazole and the first drug in the class of nitroimidazoles to have been approved for clinical use in adults. Recently, it has also been approved for use in children >6 years of age. 38 It inhibits mycolic acid biosynthesis, specifically synthesis of ketomycolates and methoxymycolates, but not α-mycolates. The exact mechanism is still not elucidated. 39 Delamanid occurs as a prodrug and its activation depends on deazaflavin (F-420)-dependent nitroreductase. Similar to the INH and ETH bioactivation, delamanid-derived intermediate metabolite has been found to form adduct with NAD+.
The adduct formation is believed to be crucial for the bactericidal effect of delamanid. 40 It has highly potent in vitro and in vivo activity against both extracellular and intracellular MTB, including replicating as well as the persister strains.4,38 A higher rate of conversion of sputum smear and culture with the improved outcome has been particularly observed in association with short-term delamanid therapy.38,41
It does not show much interaction with other drugs as neither has any effect on the cytochrome P450 enzymes nor does it get metabolized much by these enzymes. 4 Natural resistance development is less frequent with genes in the deazaflavin-dependent nitroreductase bioactivation pathway responsible for the few incidences of resistance to delamanid. 4 Overall, it has a safe profile, good tolerability, and a clinically nonsignificant effect on QT-interval (may cause prolongation).4,41
Pretomanid
Pretomanid, also named PA-824, is a nitroimidazooxazine compound that is active against replicating as well as dormant or persistent MTB strains. It is also effective for both drug-susceptible and drug-resistant MTB.4,42 It is bactericidal in nature with multiple mechanisms of action, thus making it a unique and desired novel anti-TB drug. On the one hand, it acts by disrupting the biosynthesis of mycolic acids by inhibiting the oxidation of hydroxymycolic acids to ketomycolic acids in actively replicating MTB strains.13,42 While on the other hand, it acts as a respiratory poison in dormant MTB strains. 43 Being a prodrug, it needs to be activated by a deazaflavin-dependent nitroreductase (Ddn, Rv3547).
Reduction of pretomanid releases reactive-nitrogen species such as nitric oxide, which disrupts the electron transport system (by targeting cytochrome oxidase enzymes), and thus hampers the adenosine triphosphate (ATP) synthesis. 39 After obtaining satisfactory results in phase III trials, it was approved in August 2019 by the FDA. The approval supports its use as a part of the combination regimen (BPaL regimen), which consists of bedaquiline, pretomanid, and linezolid for treatment of adult patients with MDR-TB and extensively drug-resistant TB.4,42 Few side effects including raised liver enzymes and peripheral neuropathy have been reported. 4 One of the proposed mechanisms of resistance development to pretomanid could be the loss of enzymes such as glucose-6-phosphate dehydrogenase or the dezaflavin cofactor F-420. 13
Although the present drug regimen consisting of highly efficient drugs came in the 1980s, the rapid development of resistance to these drugs along with increasing incidences of TB-HIV coinfection has limited the use of these drugs and demanded new molecules with their respective targets in MTB. Several studies are ongoing, and several of these new drugs are at different stages of development, with some in clinical trials while others are still in the experimental stage.44,45
Cell wall-acting agents with potential for therapeutic application in the pipeline:
Benzothiazinones
These are nitroaromatic-based compounds (nitrobenzothiazinones) that form a class of highly active, bactericidal, novel antitubercular drugs. 46 They perform their mycobactericidal activity at a very minimal concentration (nanomoles) by inhibiting arabinogalactan synthesis in the cell wall. BTZ-043, the leading representative molecule of this class, occurs as a prodrug, and gets activated by the flavoenzyme DprE1 (decaprenylphosphoribose-2′-epimerase). The activation results in formation of a nitroso metabolite of BTZ-043, which in turn reacts with the substrate binding site of DprE1 to cause its irreversible inhibition.
The inhibition of DprE1, in turn, prevents the synthesis of decaprenyl phosphoryl arabinose (D-arabinofuranose), an important precursor of the cell wall arabinogalactans and arabinomannans.46,47 Thus, BTZ-043 acts as suicide inhibitor. 48 These drugs are observed to be highly selective and efficacious against MTB, including both MDR and XDR strains. It also shows additive action but no antagonistic effects with most of the currently used anti-TB drugs, and has a good safety profile. 46 But benzothiazinone (BTZ) compounds have lesser activity against nonreplicating MTB strains, indicating that they need to be combined with other antitubercular drugs for effective use. 49 Resistance to BTZ-043 may occur due to the mutation at Cys387 position of dprE1 gene. 49
PBTZ169, better known as macozinone (MCZ) or the second-generation benzothiazinone, is a bactericidal compound based on piperazinobenzothiazinone. 50 Although it is similar to its precursor molecule, BTZ-0043, in terms of mechanism of activity (inhibition of enzyme DprE1), it has additional advantages over the parent molecule. First, it has been observed to be much more potent against MTB strains in experiments conducted in vitro, ex vivo, and in vivo. 50
Second, it is easily synthesized due to the absence of chiral centers and much more stable due to the presence of cyclohexyl components. 8 It has demonstrated synergistic activity with bedaquiline and clofazimine in several studies. 50 The only limitation with these candidate drugs is the lack of activity, or very poor performance to the nonreplicating or latent strains. However, the synergistic effect of PBTZ169 with other drugs tends to overcome its poor activity against nonreplicating strains. 51 Both of these novel drugs are in phase II of clinical trial studies.46,47,50
SQ109
SQ109 (1, 2-ethylenediamine) is a novel mycobactericidal candidate drug. 47 Ultrastructural analysis has demonstrated a structural similarity of this novel drug to EMB with inhibition of assembly of the cell wall complexes as its mode of activity. 52 It is an oral drug with poor water solubility that has good synergistic effect with both first- and second-line antitubercular drugs. 13 The main target of this drug appears to be MmpL3 protein. Of the 13 MmpL proteins synthesized by MTB, MmpL3 is an important transporter protein for several surface lipids (trehalose monomycolate) and proteins of the cell wall of mycobacteria, and hence is very essential for their survival and cell division.52,53
As SQ109 inhibits MmpL3 protein, the transport of surface lipids gets affected by the accumulation of trehalose monomycolates, and a deficiency of trehalose dimycolate and other cell wall mycolates. This leads to an overall mycolic acid synthesis inhibition and in turn their attachment failure with the cell wall arabinogalactans. 52 The spectrum of action of SQ109 includes mycobacterial strains implicated in causing MDR as well as XDR-TB. Although structurally similar to EMB, they show good activity against MTB strains that are resistant to the former. 47
Some studies have demonstrated their in vitro synergy with some of the first-line antitubercular drugs such as INH and rifampicin, with their activity even against rifampicin-resistant strains of MTB. It also shows an additive action with streptomycin and bedaquiline. 54 At present, this compound is in phase II clinical trials. 47
Meropenem–clavulanate
The β-lactam group of antibiotics has served as a boon for an extensive period as they have a notable bacteriostatic action against a variety of Gram-negative and Gram-positive bacteria species. MTB is observed to be having intrinsic resistance to this group, probably due to the poor permeability of the outer membrane and the presence of a highly potent β-lactamase BlaC. The BlaC is a member of Ambler class A β-lactamases, which is sensitive to almost all β-lactamase inhibitors. 55 Hence, this combination of β-lactam and β-lactamase inhibitors may act as good targets even for TB. Some recent studies have shown that meropenem (relatively poor/weak substrate for BlaC)–clavulanate combination can overcome the problem of intrinsic resistance in MTB, thus acting as an important anti-TB candidate.
The in vitro studies have shown good activity of this combination against both drug-susceptible and MDR-TB and XDR-TB strains. 56 The combination is also effective against nonreplicating MTB strains. 57 Meropenem exerts its mycobactericidal action by inhibiting the enzyme L,D transpepetidase that catalyzes the 3 → 3 crosslinking in the peptidoglycan layer. 58 On the contrary, faropenem, an oral carbapenem used for TB treatment, has been shown to arrest the growth of MTB instead of lysing the cells. Thus, there is an emerging debate on revisiting the actual mode of action of β-lactam antibiotics on mycobacterial cells. 59
Cell wall-acting agents in the experimental stages
Capuramycin
This is a naturally occurring antibiotic that has a uracil nucleoside structure with a caprolactam substituent. These were isolated from the culture filtrate of Streptomyces griseus 446-S3. 8 The native capuramycin has been named SQ997, while two of its important analogs are SQ641 and SQ922, of which SQ641 is the most potent compound against all Mycobacterium spp. 60 SQ641 has also demonstrated good synergistic effect with other antitubercular drugs and postantibiotic effect >48 hours. 60
Mycobacterial translocase I (MurX/MurY), an enzyme involved in peptidoglycan cell wall synthesis, is the main target of capuramycin and its analogs. 61 These analogs have been observed in some studies to be rapidly bactericidal. One of these analogs UT-01320, which also targets bacterial RNA polymerases, has been observed to be bactericidal for even the dormant TB bacilli at low oxygen concentrations. 61 Although the in vitro studies have demonstrated these compounds to be of good potency, their low solubility in water limits their proper absorption when given orally and thus their use in vivo.8,60 Various drug delivery vehicles for the same are still under study. 8
Teixobactin
A novel naturally derived antimicrobial that has been described recently, produced by a new species of soil proteobacteria, named Eleftheria terrae. 62 It is a cyclic depsipeptide containing an unusual amino acid enduracididine. 63 It shows excellent bactericidal activity against a wide range of drug-resistant Gram-positive bacteria such as Staphylococcus aureus, Streptococcus pneumoniae, and M. tuberculosis. There is also moderate-to-good activity against Clostridioides difficile and Bacillus anthracis. 63
It has a novel mechanism of action, which is different from that of other cell wall inhibitors as it acts on targets that are not protein by nature. These targets include a highly conserved motif of lipid II (precursor of peptidoglycan) and lipid III (precursor of teichoic acid).62,64 One of the limitations to its broad-spectrum clinical use is its poor oral bioavailability. 65
Other molecules in the preclinical stages have been summarized in Table 2.66–73
Potential Compounds Targeting Mycobacterial Envelope in the Experimental Stage
TLM, thiolactomycin.
Compounds acting on the mycobacterial cell membrane
The mycobacterial cell membrane can also serve as an important target for the development of new drugs in both actively dividing and nondividing, latent cells as it takes part in many of the critical processes necessary for mycobacterial cell survival. 12 Most of these drugs are at the experimental stage and require further studies. The important drugs being considered recently against MTB are mentioned here.
Antimicrobial agent
Boromycin (named because of the presence of boron) is a polyether-macrolide compound. It was originally isolated from a fermentation broth of Streptomyces spp. A-3376.74,75 It was found to be a strong anti-HIV antiviral agent that inhibited the replication of both clinical isolates and laboratory-cultured isolates of HIV-1. Among the bacterial population, it is observed to be active against Gram-positive organisms and ineffective against Gram-negative strains due to the presence of lipid-rich outer cell membrane in the latter. Acting as an ionophore for potassium ions, it breaks the transmembrane potential of the mycobacterial membrane, thus depleting the intracellular levels of ATP and causing lysis of the cell releasing all its contents to outside. 74
Repurposed drug
Verapamil has been a compound of recent interest for its efficacy to potentiate treatment options for drug-resistant TB and to reduce the total time required for their treatment. It acts as a calcium channel blocker, and has been used clinically for hypertension and other cardiovascular indications. Verapamil shows antimycobacterial activity in a dose-dependent manner mainly due to inhibition of the membrane efflux pumps, which concentrates high levels of anti-TB drugs within the organism causing its death. 76
Verapamil is cationic amphiphilic in nature. It has been observed in some studies that concentration-dependent loss of the mycobacterial membrane potential occurs on priming the bacteria with this drug before giving anti-TB treatment. Both verapamil and norverapamil augment the in vitro activity of some novel anti-TB agents such as bedaquiline and clofazimine on both drug-susceptible and resistant strains. 12 MTB-specific CD3 T cell expansion is also significantly suppressed by verapamil and norverapamil.
Recently, a thorough search for new analogs having better efficacy is underway. Among the new analogs discovered, effects of three, namely, KSV21, MKV4, and MKV9, have been studied on intracellular BCG, with KSV21 having maximum potency. 76 These analogs have the benefit of potentiating the inhibitory activities of INH and rifampin too on intracellular MTB, and do not interfere with the replication and expansion of MTB-specific T cells.12,76
Antimicrobial peptides
The endogenous cationic antimicrobial peptides (AMPs) formed as defense peptides in hosts are amphipathic molecules that target the cell membrane of the microbes and disrupt it by forming toroidal, barrel stave, or carpet-like pores.77–80 They are also immunomodulatory, thus acting as important anti-infective agents. 81 It has also been observed that the accumulation of ubiquitin-derived peptides (a synthetic ubiquitin peptide—Ub2) in the lysosomes enhances its bactericidal activity against mycobacteria by altering its cell membrane stability. 82
Plant products
Xanthones (a group of biological compounds having great structural diversity) occur both naturally in plants and obtained by chemical synthesis. 83 Initially, some xanthones were reported to be having good antibacterial activity against a few Gram-positive bacteria, but they could not be evaluated for clinical use. Later on, with some modifications, new, improved, and more potent analogs of these compounds were developed having selective activity against some particular pathogens, which also included MTB.
Three major analogs have been developed of which analog 1 (AM-0016) is observed to be most potent against MTB. 84 Its activity profile has been studied on Mycobacterium bovis cultures. AM-0016 destroys the mycobacterial cell envelope by causing disruption in the membrane potential, and increasing its permeability to other drugs and toxins that are harmful to the cell viability, thus making it bactericidal by nature. The positive aspects of its activity are its rapid action even on the nonreplicating, latent bacterial cells and a low propensity to develop resistance. 12
Other compounds
Some other compounds with the potential of targeting mycobacterial membrane have also been identified from the indolyl-mannich bases and the indolylalkyltriphenylphosphonium bases with their analogs, which have the cationic amphiphilic properties. The triphenylphosphonium TPP cation has already been a known membrane targeting molecule. The addition of this cation to some compounds having activity against MTB can potentiate the activity of that drug by increased intracellular accumulation of that compound. All these candidate drugs in general have been observed to be degrading the membrane potential, and hence the permeability of the cell membrane. 12
Challenges in the Development of Drug Molecules Targeting Mycobacterial Cell Wall
With the increasing number of cases of MDR-TB, there had been a great need and pressure for the development of new antimycobacterial drugs. Although multiple compounds acting on the mycobacterial cell wall are in the clinical/preclinical trials or the experimental stage, there are several hurdles in their way to becoming successful candidates for therapeutic application in mycobacterial infections.
Variations in the clinical strains
MTB is a highly complex bacterium with substantial biological differences among various clinical strains, which affect not only its virulence and overall disease progression but also its survival, response to treatment with antimycobacterial drugs, and in turn the development of antibiotic resistance. 85 For the process of development of new drugs against MTB, these challenges of genotypic and phenotypic diversity are to be well known, well studied, and overcome efficiently. The variations in MTB can be discussed under two broad categories, the genotypic or genetic variations and the phenotypic variations, which may include the variations in the cell wall composition and variations in susceptibility profile of MTB. 85
Genetic/genotypic variations
Although it was believed earlier that MTB has a relatively conserved genomic environment, recent techniques such as next-generation sequencing have revealed significant genetic diversity, particularly about the genes required for basic survival phenomenon like growth and metabolism. 86 It has also been observed that certain genes are strain specific. Few studies have identified some genes such as katG and glcB whose occurrence in a particular strain defines the treatment response of that particular strain to the antimycobacterial antibiotics. 86 Also, the genetic variance and their single nucleotide polymorphisms that occur due to faulty DNA replication or repair are mainly responsible for the development of resistance in MTB, posing a major challenge in developing new therapeutic agents. 87
Variations in cell wall composition
While the majority of new drugs in the experimental/clinical trial stage are based on the targets present in the cell wall of mycobacteria, several MTB isolates have been observed to possess considerable differences in the composition and alignment of various components of their cell wall. Some studies outline these differences by comparing multiple strains of varied lineages of the MTB complex.
For instance, East-Asian and Euro-American lineages have some peculiar lipids in their cell wall, and thus might show high virulence, transmissibility, and pathogenicity, while the Ethiopian lineage lacking those compounds shows less virulence and low transmissibility. 88 This can be explained based on the fact that lipids in association with other cell wall components form a tough hydrophobic layer that protects the bacterial cell from the stresses offered by the host immune system as well as the antibiotics. 89
Another factor responsible for cell wall variation among different strains of mycobacteria is the asymmetrical cell division as well as the asymmetrical deposition of newly synthesized cell wall components at the tips of the bacterial cell. Asymmetrical cell division occurs due to LamA (loss of asymmetry mutant) gene present in the dividing mycobacterial cells.
The major role of LamA is to relocate and concentrate at the dividing septum, and in association with other components of the cell wall inhibits the growth of newly developing pole resulting in gross asymmetry. Thus, the daughter cells become phenotypically heterogeneous, with respect to their cell size, their virulence, and their response to drugs. Also, various cell wall components such as proteins, lipids, peptidoglycan, arabinogalactan, mycolic acids, and even the enzymes required for cell division are mostly concentrated toward the growing poles (particularly old poles) or division septa. 11
Variations in susceptibility profile
This challenge is seen specifically for the beta-lactam and beta-lactamase combination drugs that are in pipeline for TB therapy. It has been observed that isolates belonging to different lineages of MTB show differences in their susceptibility to this antibiotic combination. A recent study by Cohen et al. has demonstrated that all the tested strains of MTB used in their study were susceptible to meropenem–clavulanate but astonishingly, ∼45% of the tested strains, many of which were MDR and XDR strains, were also found susceptible to amoxicillin/clavulanate.
This observation was surprising because apart from the meropenem–clavulanate combination, MTB, in general, is resistant to the beta-lactam group of antibiotics. It was also observed by analysis that those strains that were found susceptible to amoxicillin/clavulanate belonged to a LAM4 clade (which was responsible for the XDR outbreak of 2005). 90 Further studies are required to decide whether this combination (amoxicillin/clavulanate) is really useful for the MDR/XDR strains as this combination can be a better alternative to meropenem–clavulanate in terms of its oral availability and safety profile.
Inactivity of the drug targets during stasis
Infection with MTB includes not only the actively replicating and growing MTB cells but also some cells that are stationary or static. These stationary cells help them to persist, thus establishing the latent TB infection and making the treatment of MTB infection lengthy and complex. 91
Although the stationary phase is a period of comparative inactivity, numerous cell wall and membrane changes happen continuously, which might have an effect on the response produced by currently available antimycobacterial therapeutic drugs and also pose a challenge to the development of new and effective drugs. Having a thorough knowledge about those targets that might be active in this persistent stage of MTB and the underlying mechanisms can provide useful guidance regarding the development of these new drugs.11,58
Along with the common lipids seen in all bacterial cell membranes, the mycobacterial cell membrane contains some special compounds such as lipomannans (LMs), lipoarabinomannans (LAMs), and their precursor molecule phosphatidylinositol mannosides (PIMs). Not only are they the key factors for maintaining cell wall integrity, but they also play important role in the establishment and progression of the disease. 11 Although they can be explored as useful drug targets for new, experimental drugs, yet the observation that they get downregulated during the period of stasis poses a major challenge to new drug development.
The universal stress protein (USP) whose activity increases (due to highly active regulator proteins such as PknG and DosR) during the period of stasis is believed to be retarding the transport and activity of PIM. Reduction in the quantity and activity of PIMs in the cell membrane of MTB during stasis causes difficulty in the development of drugs that might use PIMs as their targets.
Contrary to this observation about PIMs, the LMs and LAMs are seen to be having a tremendous increase in their both quantity and activity, during the static or latent phase. This is probably due to an increase in the activity of the genes that code for enzymes involved in the synthesis of LMs and LAMs (example includes up regulation of embC gene). The increased concentration of LM and LAM in the latent phase helps in evading the immune response and probably the antibiotic therapy, thus maintaining the infection. 11
The individual layers of the cell wall and their components show a difference in their activities during the stationary phase, and this property will influence the susceptibility or resistance response of MTB to antibiotics.
For the peptidoglycan layer, the deciding factor for its response to antibiotics in the stasis period is the alteration in the activity of PknB, a protein kinase acting as the regulator of peptidoglycan synthesis and metabolism. During the stationary phase or any sort of stressful environment, PknB gets inhibited, which in turn downregulates peptidoglycan metabolism, making the cell wall more resistant toward the entry of antibiotics. 11
Some transpeptidases (which crosslink the peptidoglycan) have been suggested as promising targets for new drugs against MTB. Among them, PonA1 and PonA2 play roles at different periods of the cell cycle with PonA1 being more active during the replicative or growth phase of MTB, while PonA2 being higher in concentration during the stasis period. Therefore, these enzyme complexes show the difference in their sensitivity patterns, with some tolerance to a few cell wall-acting agents.11,92
Several genes play roles in arabinogalactan synthesis and metabolisms such as rmlA, rmlB, and rmlC. Some of these get inhibited by pknG, which is increased during stasis. Thus, the arabinogalactan synthesis and metabolism are downregulated in the stationary phase. Also, the mycobacterial arabinotransferases or the Emb proteins such as EmbA, EmbB, and EmbC get differentially regulated between the growth phase and stasis phase. Those that predominate in arabinogalactan synthesis such as EmbA and EmbB get upregulated during the growth phase, while those that are involved mainly in LAM synthesis such as EmbC are accumulated during the stasis phase. This again leads to increased LAM synthesis, which alters antibiotic tolerance of the mycobacterial cells.11,93
The fatty acid synthases (FASs—FAS I and FAS II) play a crucial role in the synthesis of mycolic acids. The FAS II enzyme helps in the production of meromycolic acids for the bacterial cell wall. 94 The FAS II enzymes are all downregulated by the enzymes PknA and PknB (which are associated with the stationary condition), thus inhibiting the mycolic acid synthesis. 11 Moreover, there is downregulation of the cis/cismeromycolates during the static phase due to which the level of oxygenated meromycolates rises. These oxygenated meromycolates promote old and new drug tolerance to the antibiotic.11,94
Pharmacokinetic properties of the molecule
One of the major challenges for a new drug development had always been its pharmacokinetic properties, defined as how the drug behaves in vivo, particularly inside the human body. Thorough analysis of these properties of a new drug as well as their comparison with those of the already existing ones is absolutely essential to assess its potency, efficacy, and superiority over the drugs already in use. An unfavorable pharmacokinetic profile may shadow approval of a new drug for use in vivo even though it shows high potency against MTB in vitro.8,95
One of the greatest challenges faced in the treatment of TB particularly for MDR-TB or XDR-TB is the oral bioavailability of antimycobacterial drugs. As we already know that many of the current second-line drug regimens for MDR-TB are injectables, which are to be given daily for ∼12–18 months, this drastically reduces the compliance of patients to antitubercular therapy, making them more prone for the development of XDR-TB.
Also the orally available drugs are always more welcomed by patients than the injectables, thus improving the therapeutic profile. Therefore, among the pharmacokinetic properties, the first and foremost challenge is analysis of the chemical nature of the drug, which includes its water solubility and oral bioavailability profile and the factors affecting them. This in turn helps deciding its correct route of administration. For instance, among the drugs already used it has been observed that their absorption and thus bioavailability might be altered by the diet of the patient. 8
Saktiawati et al. have shown in their study that food lowered the maximum concentrations of INH, rifampicin, and pyrazinamide by 42%, 22%, and 10%, respectively, while the concentrations of EMB were not affected. Also the time to maximum concentration was delayed for INH with a high-fat meal. 96 On the contrary, the newer compounds such as delamanid and pretomanid are seen to have a markable enhancement (two-to-fourfold) of bioavailability by food. 97
It is well known that TB is a disease with a wide spectrum of presentation, and it may involve any of the organ systems. Therefore, the anti-TB regimen should include drugs, which can have enhanced volume of distribution and penetration into various areas of the body. Thus, the second challenging pharmacokinetic property is that the volume of distribution of drug depends on the chemical composition of the drug as well as its protein binding capacity. 8 It has been observed that the hydrophilic drugs may not penetrate hydrophobic areas of the body.
Similarly a highly protein-bound drug will have a lower volume of distribution and vice versa. These properties play a crucial role in treatment of certain challenging cases of TB such as TB meningitis. For example, some of the anti-TB drugs have remarkable penetration in cerebrospinal fluid, such as INH (hydrophilic), cycloserine, and delamanid, while others such as EMB, though, have a good and rapid volume of distribution they have poor cerebrospinal fluid (CSF) penetration (CSF penetration only in the presence of inflammation). 98
On the contrary, certain drugs may have a restricted entry to other TB-affected areas of the body such as INH, and may not penetrate or accumulate in caseous granulomas. 99 This suboptimal drug penetration at different sites of infection may lead to persistence of bacteria within host and in turn to rapid development of resistance even to a newly discovered drug. 100
The third challenge encountered is the activation of the inert drug molecules referred to as prodrugs. Prodrugs are pharmacologically inactive compounds that need to get metabolized by particular enzymes of the host cells to the active drug moiety, which in turn shows antibacterial action. There are several antimycobacterial drugs, which exist as prodrugs and require activation in vivo as the drugs already in use such as INH, ETH, and among the newly studied and approved, delamanid and pretomanid.8.101
Prodrugs can act as both a boon and a curse for antimycobacterial therapy. These are inert compounds. Therefore, they do not lose their activity while absorption or transportation to the target cells, which leads to several advantages such as better solubility, bioavailability, and longer half-life leading to enhanced and targeted drug delivery, which improves their therapeutic efficacy against MTB and reduces adverse effects. 101 On the contrary, if the host is lacking the enzymes required for their activation, then these compounds would fail to exert their antimycobacterial action, and instead may contribute to development of MDR or XDR-TB despite optimal administration of therapy.
Another important aspect of pharmacokinetics is the metabolism of the drugs inside the host, which also defines its efficacy. As an example, we can observe the process of acetylation by which several drugs are metabolized in the host. One of the most important antimycobacterial drugs INH loses its potency due to such action. Acetylation occurs by a hepatic enzyme N-acteyltransferase and based on the rate of acetylation, the human population is divided into two main phenotypic groups, fast and slow acetylators.
Studies have shown that in comparison with the fast acetylators, slow acetylators have higher INH bioavailability, longer duration of bactericidal levels of the drug in the blood, and slower elimination of the drug from the host. 102 Therapeutic drug monitoring may be used as an important tool in such cases to establish appropriate dose for individual patients. 99
As for the β-lactam group of antibiotics, due to the presence of the β-lactamase enzyme BlaC in the mycobacterial population, a higher dose of the carbapenem antibiotics or their combination with β-lactamase inhibitors is necessary. Also, for extracting maximum activity, therapy with divided doses has been suggested. 99 Although the metabolism of any drug is dependent upon an individual host and is genetically determined, yet this is an important challenge that should be looked into while searching and experimenting with new drugs. 102
Finally, the pharmacokinetic factor to be taken into consideration is the elimination or excretion of the drug from the body. This factor may act as an important challenge in new drug development as it largely depends upon the chemical nature of the drug, wherein the hydrophilic drugs such as INH undergo direct and easy excretion without much metabolic changes while certain hydrophobic drugs such as delamanid and pretomanid have to undergo some metabolic modifications to convert them into polar molecules for ease of their excretion. 103
As the major organ of excretion is kidney for most of the drugs, followed by intestinal elimination for a few of them like delamanid therefore, any disease or dysfunction affecting these excretory organ systems will also affect the elimination process and in turn may be a significant cause of severe toxicities. This has been well observed in the patients with renal compromise as they show high incidences of retrobulbar neuritis with a high-dose EMB containing regimen.99,104 This may act as a major concern particularly for the upcoming drugs having a narrow safety window.
Pharmacodynamic properties of the molecule and drug toxicities
Pharmacodynamics, in general, refers to the overall effects of a drug on the body of the host or any living system. It is an important aspect to look into as it not only gives information about the therapeutic effects and toxicities of the drug under study but also provides insights into the possible interactions between different drugs that might be prescribed or taken together. 105 The safety profile of a drug and its associated toxicities may be deciphered from the clinical trials.
The knowledge of the toxicities of various upcoming drugs is extremely important as these adverse effects may be quite serious, and thus restrict the therapeutic use of the drugs. For instance, thiacetazone (TAC), one of the oldest and a highly potent drug acting on mycobacterial cell wall (inhibits the mycolic acid synthesis) has been removed from the antitubercular regimen owing to a plethora of toxic effects, including bone marrow depression with anemia, leukopenia, agranulocytosis, thrombocytopenia as well as red cell aplasia, hemolyticanemia, and hepatotoxicity with jaundice. 106
Similarly, patients with low renal creatinine clearance are to be treated with longer interval doses of EMB containing regimen, as frequent or higher doses may lead to drug accumulation and retrobulbar (optic) neuritis. Patients with preexisting cardiac conditions have to be monitored carefully while treating with delamanid as it causes cardiac QT prolongation due to its long half-life. Therapeutic drug monitoring is emerging as a new and important tool to overcome these challenges with metabolism and excretion of the drugs, and thus may boost up the new drug discoveries. 99
Drug–drug interactions
The anti-TB treatment faces a major challenge with HIV coinfection. Since TB is the most common and frequent opportunistic infection seen at advanced stages of HIV, the problems arising as a result of this coinfection are to be dealt with at every step. Each of these diseases alters different parameters of the other disease such as the clinical presentation, diagnostic evaluation, and therapeutic modalities with the most common problem being the anti-TB drug and antiretroviral drug interactions. These drug interactions not only affect the existing panel of available drugs but also act as a challenge in the development of new drugs for either of the diseases. 107
Some of the antiretroviral drugs interfere with the metabolism and therapeutic levels of anti-TB drugs. This is seen particularly for the newly developed and approved anti-TB drugs such as bedaquiline, delamanid, and pretomanid. It has been observed in various studies that when efavirenz is given along with bedaquiline, it decreases the blood concentration of bedaquiline, reducing its efficacy while ritonavir-boosted protease inhibitors increase its blood concentration by reducing its elimination. 6 Similarly, efavirenz, protease inhibitors, and some anti-TB drugs such as rifampicin decrease the blood levels of pretomanid.99,108 No such interaction is seen with delamanid and antiretroviral drugs, and thus they can be used safely together. 6
Resistance mechanisms to new drugs
First-line antimycobacterial drugs were great discoveries for the treatment of TB that had grossly affected mankind for ages. But with the great discovery came the greater problem. The development of resistance has become inevitable now with the mycobacteria having the potential to develop resistance mechanisms to even the newly approved drugs. Although mycobacteria show intrinsic resistance to some drugs by the virtue of their nonpermeable cell wall, the most common mechanism of acquired resistance in a previously sensitive mycobacterial strain is the occurrence of mutations in the genes that encode for the targets of the antitubercular drugs.
Moreover, the noncompliance to antimycobacterial therapy with on and off treatment leads to selective replication of these mutants, which ultimately converts a drug-susceptible mycobacterial strain to a drug-resistant strain. This mechanism is observed for almost all the first-line anti-TB drugs. 109
The inherent tolerance to a certain group of antibiotics is due to the high lipid-containing molecules of the cell wall such as LMs, LAMs, and their precursor molecule PIMs. Moreover, the presence of enzymes such as β-lactamases adds to their resistance pattern for an important group of antibiotics; that is, β-lactams. Still, the combination regimen of meropenem and clavulanic acid is studied for its antimycobacterial activity. 8
Studies have observed certain mutations, which might be associated with resistance to the novel antitubercular drugs that have been either recently approved or in the pipeline (mentioned under the description of specific drugs). 110 Therefore, there is an urgent need to develop new methods to detect these resistance patterns in the novel drugs at the earliest.
Challenges in the Development of Drug Molecules Targeting Mycobacterial Cell Membrane
The limitations and challenges encountered with cell membrane-acting agents may be slightly different as compared with the already approved drugs and the drugs acting by a different mechanism. Although overall a better knowledge is always helpful, these novel drugs will require more specific knowledge regarding their mechanism of action, the side effects, and their safety profile to be later approved for their role in the treatment of mycobacterial infections.
One of the major limitations of this group of drugs would be their lack of selectivity for the mycobacterial cell membrane. The mammalian cell membranes are also made up of lipid bilayer with some of the similar kinds of lipids as observed in the mycobacterial cell membrane. Also unlike the bacterial cells, the mammalian cell membranes are not protected by a rigid cell wall, thus they are more prone to damage from any stress factor, whether physical or chemical in the form of drugs.
Some of the novel drugs are attracted to lipid membranes of the mycobacteria as well as the mammalian cells. While damaging the mycobacterial cell membranes, the drugs may also damage the membranes of the host cells. There is a recommendation stating that these novel compounds should have a 10-fold selectivity against mycobacterial cells in contrast to mammalian cells. 111 Although most of these drugs preferentially act upon bacterial membranes, some of the compounds, particularly the small-sized molecules, may target the mammalian membrane. 112
Some studies have observed this phenomenon with an AMP that caused some lipid storage disorders in the host. In vitro experiments in the form of cytotoxicity assays with mammalian red blood cells and in vivo studies for understanding the safety range and toxicities due to the novel drugs should be undertaken to overcome this challenge. 12
Another challenge faced with these drugs is the lack of thorough understanding of the specific process of their action. Studies have observed that some of these drugs cause cell membrane depolarization but do not alter the permeability of the membrane, while some others alter both the membrane potential and the membrane integrity, thus disrupting the impermeable barrier. This ambiguous observation is a big challenge and requires a clear understanding of the detailed mechanism of action of these membrane-acting drugs. 12
Constraints in the Evaluation of the Promising Drug Molecules Against Mycobacterial Cell Envelope as Effective Therapeutic Candidates
Apathy for translational research
It would not be wrong to state that actual interest in TB and its treatment modalities came up when HIV had hit the entire world badly with TB-HIV coinfection rising at an alarming rate. At the very same time, the drug-resistant TB cases came into realization along with the failure of the existing drug regimen. The belief that TB is not a threat for the developed nations, but a disease of the developing and poor nations had shifted the focus of the major part of the world toward other communicable and noncommunicable diseases.
Consequently, there had been inadequacy in research-related investments that ultimately led to the paucity of translational researches and clinical studies for newer effective drugs against MTB. As the MDR-TB cases were observed to be rising at a tremendous rate, there was a reshifting of interest in the treatment of TB. It was only by 1993 that the WHO took interest in TB and declared it as a disease of global concern. Thereafter, the search for promising drugs and their clinical trials were expedited. This lack of concern has led to a substantial delay in the development, testing, and approval of new antimycobacterial molecules, and will require more sincere and dedicated efforts for the discovery and testing of novel drugs. 8
Impediments to preclinical and clinical trials
Preclinical and clinical trials are essential parts of any new drug development as they help in the assessment of the efficacy, the spectrum of activity, dosing, route of administration, interactions with other drugs, and the safety profile of the drug under study. A novel drug cannot be approved for clinical use unless some satisfactory results have been obtained from its trial. The trials for novel antimycobacterial drugs including those acting against cell envelope were posed with many challenges and limitations.
- Lack of well-known, easily measurable, and reliable prognostic biomarkers to indicate the success of treatment with novel drugs.
- No genuine animal models for MTB are available for preclinical experiments and studies, which can predict the outcome of treatment with new drugs.
- In resource-limited countries with huge populations and a high burden of TB and TB-HIV coinfection, there is paucity of the proper sites for the trial as well as of the informed and concerned volunteers who can help make these trials successful.
- As the treatment for TB requires long periods of observation, these trials are usually very lengthy and cumbersome. This not only raises several ethical issues regarding the trial and its participants but also reduces the interest and compliance of the participating volunteers.
- Due to the ever-increasing frequency of development of resistance even in the newly discovered drugs, pharmaceutical companies and investors refrain from funding for the novel drug trials for the fear of a reduced return of profit.
Footnotes
Acknowledgments
The authors thank researchers and scientists across the globe for working relentlessly for developing antitubercular drugs.
Authors' Contributions
A.D. planned the article, and A.M. performed the literature search. Both A.M. and A.D. prepared the initial draft. A.D. designed the figures. A.D. and T.B. edited and critically evaluated the final draft.
Disclosure Statement
We have no conflict of interests to declare.
Funding Information
No funding has been received for writing this article.