
Abstract
Salmonella is a foodborne zoonotic pathogen and a hazard to public health. Surveillance of the prevalence of Salmonella is important. This study sought to understand the population structure, antimicrobial susceptibility, and virulence-associated gene profile of 100 Salmonella, which were randomly selected from clinical foodborne diarrhea fecal samples during 2015 and 2019 in the Jiangsu Province, China. After whole-genome sequencing and in silico analysis, we found that the prevalence of clinical foodborne Salmonella in Jiangsu Province was periodic and that the serotypes were diverse, covering 9 serogroups and 19 serotypes. S. Enteritidis was the most prevalent serotype, followed by S. Typhimurium. A high prevalence of antimicrobial resistance was also observed in this study, nearly half (47/100) of Salmonella isolates were determined to be multidrug-resistant (resistant to ≥3 antimicrobial agents), the antimicrobial resistance genotype and phenotype were associated but not closely related, and antimicrobial resistance differed between the major Salmonella sequence types. Additionally, we found that the virulence-associated gene profile is highly concordant with the serotype. Our work shows the association among serotype, antimicrobial resistance, and virulence gene profile, demonstrating the connection between genotype and phenotype and providing epidemiological data for Salmonella.
Introduction
Salmonella is a gram-negative zoonotic foodborne pathogen and a major hazard to public health in many countries. 1 There are two species that belong to the Salmonellae genus, S. bongori, and S. enterica. The major subspecies of S. enterica, S. enterica subsp. Enterica includes more than 2,600 different serotypes that can cause diseases in a wide range of hosts.2,3 Human infections by Salmonella are commonly associated with food products of animal origin, such as meat, eggs, and milk. In China, Salmonella is responsible for approximately 70–80% of foodborne pathogenic disease outbreaks. 4
Generally, antibiotic treatment is an easy and effective way to treat bacterial infections. 5 However, with the abuse of antimicrobial agents, a series of multidrug-resistant bacteria have emerged.6,7 According to a report by the Chinese Academy of Sciences, China consumed approximately 162,000 metric tons of antibiotics as of 2013. 8 Because Salmonella infection is usually a self-limiting disease and generally recovers on its own within one week, it is not recommended to use antibacterial drugs for treatment. 9 However, Salmonella infection carries a high mortality rate in immunodeficiency patients, and in these cases, antimicrobial agents are crucial for treatment. The antimicrobial resistance of Salmonella is becoming a serious threat to public health. 10
At present, there is no report on the use of whole-genome sequencing (WGS) technology to explore the prevalence and drug resistance of Salmonella in Jiangsu Province. In this study, we randomly selected 100 Salmonella isolated from the feces of diarrhea patients in Jiangsu Province, China, during the period from 2015 to 2019. We assessed the demographic characteristics of patients, serotype distribution, phylogenetic characteristics, antimicrobial resistance genotypes, and phenotypes of bacterial isolates by using WGS data. Based on these results, we aim to deeply explore the epidemic and drug resistance characteristics of Salmonella in Jiangsu Province. This will provide a basis for the monitoring, early warning, prevention, and control, as well as treatment of Salmonella in this region. Moreover, it may also offer new ideas and directions for the development of Salmonella vaccines.
Materials and Methods
Bacterial strains and cultures
Salmonella isolates and demographic information were collected from patients with a diagnosis of salmonellosis from hospitals in the Jiangsu Province, China between 2015 and 2019. A total of 100 Salmonella isolates (20 per year) were randomly selected and analyzed in this study. These isolates were purified using MacConkey and Salmonella and Shigella (SS) plates. All Salmonella isolates used in this study were cultured in Luria-Bertani broth and are listed in Supplementary Table S1.
Whole-genome sequencing
Bacterial genomic DNA was extracted using the FastPure Bacterial DNA Isolation Mini Kit (Vazyme, China) following the manufacturer’s instructions. WGS was performed on the Illumina NovaSeq PE150 platform (Illumina, https://www.illumina.com), followed by de novo assembly using SOAP denovo (version 2.04).11,12 Gapclose (Version: 1.12) was used to optimize and fill the gaps.
Serotyping and MLST
SeqSero 1.2 (https://cge.cbs.dtu.dk/services/SeqSero/) was used to predict the serotypes of the Salmonella isolates. Seven housekeeping genes, aroC, dnaN, hemD, hisD, purE, sucA, and thrA were used to perform multilocus sequence typing (MLST), 13 and pubMLST (https://pubMLST.org) was used to determine the sequence type. PHYLOVIZ was used to visualize Clonal Complex (CCs) and to create an MLST-based minimal spanning tree following a goeBURST algorithm.
Phylogenetic analysis
To analyze the homology of the Salmonella isolates, Roary software was used to align the core-genome of these Salmonella isolates, 14 and a phylogenetic tree based on core-genome alignments was constructed using RAxML using the general time reversible plus GAMMA distribution substitution model. 15
Antimicrobial susceptibility phenotype and genotype analysis
Antimicrobial susceptibility of these Salmonella isolates was tested following Clinical & Laboratory Standards Institute (CLSI) 2017 using VITEK® 2 COMPACT (bioMérieux, USA). Salmonella isolates that demonstrated resistance to at least three different antimicrobial agents were defined as multidrug-resistant isolates. 16 The following antibiotics were used in this study: ampicillin (AMP), trimethoprim/sulfamethoxazole (SXT), imipenem (IPM), tetracycline (TET), gentamicin (GEN), chloramphenicol (CHL), cefotaxime (CTX), ceftazidime (CAZ), cefoxitin (CFX), ciprofloxacin (CIP), nalidixic acid (NAL), and azithromycin (AZM).
ResFinder 4.1 was used to identify the acquired antimicrobial resistance genes (ARGs) and chromosomal mutations based on WGS data.17,18
Virulence-associated gene profiling
The virulence-associated gene profiling of the Salmonella isolates was extracted from WGS data by using a VFanalyzer. 19 Virulence factors (VFs) were classified by their function and role in pathogenesis, such as coding for capsular proteins, fimbrial adherence determinants, and secretion systems. Genes (clusters) that were homologous with known VFs in other bacterial pathogens were also scanned, potentially elucidating the genetic shift between different bacteria in evolution.
Ethics statement
This study was approved by the ethics committees of the participating local hospitals and the ethics committee of the Chinese Centers for Disease Control and Prevention.
Results
Demographic characteristics of Salmonella infection patients
In this study, we randomly selected 100 Salmonella isolates from patients with a diagnosis of salmonellosis in the Jiangsu Province, China, between 2015 and 2019. The case reports and WGS data were collected. Fifty-six percent of these Salmonella isolates were isolated from male children (aged 0–14 years) (Fig. 1A). Patients ≤3 years of age made up 44% of our sample population while other age groups were evenly distributed, indicating that children are more susceptible to salmonellosis (Fig. 1B).
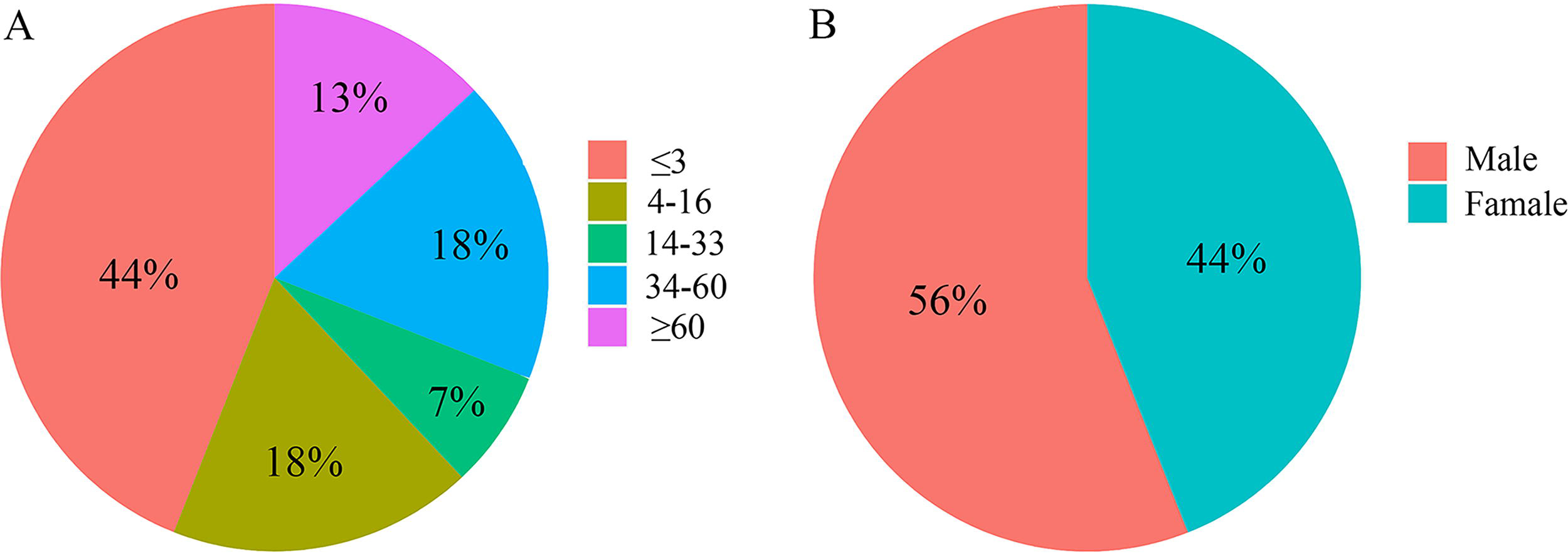
The distribution of Salmonella isolates sourced by
The population structure distribution of Salmonella
All serotyping results were derived from SeqSero2 and a diverse serogroup was covered, including A, B, C1, C2, C3, D, E1, E4, and F. Among these serotypes, S. Enteritidis (35%) and S. Typhimurium (34%) were the most predominant. S. Enteritidis comprised only ST11 and S. Typhimurium comprised 4 sequence types (ST); more than half (20/34, 58.82%) of these STs were ST34, followed by ST19 (12/34, 35.29%). S. Thompson was the third most common serotype in this study (7%), and the majority was ST26 (6/7, 85.17%). Other serotypes were detected in only a limited number of cases (Fig. 2).

Sequence type (ST) distribution of Salmonella in relation to serotype. N/A, not available in SeqSero.
Through phylogenetic analysis of the core-genome of these Salmonella isolates, we found that the Salmonella isolates belonging to the same serogroup or serotype were closely related in the phylogenetic tree. This was especially true for serogroups B and D which were mainly composed of S. Typhimurium and S. Enteritidis, respectively, indicating that the serogroup/serotype of Salmonella was closely related to core-genome evolution (Fig. 3).
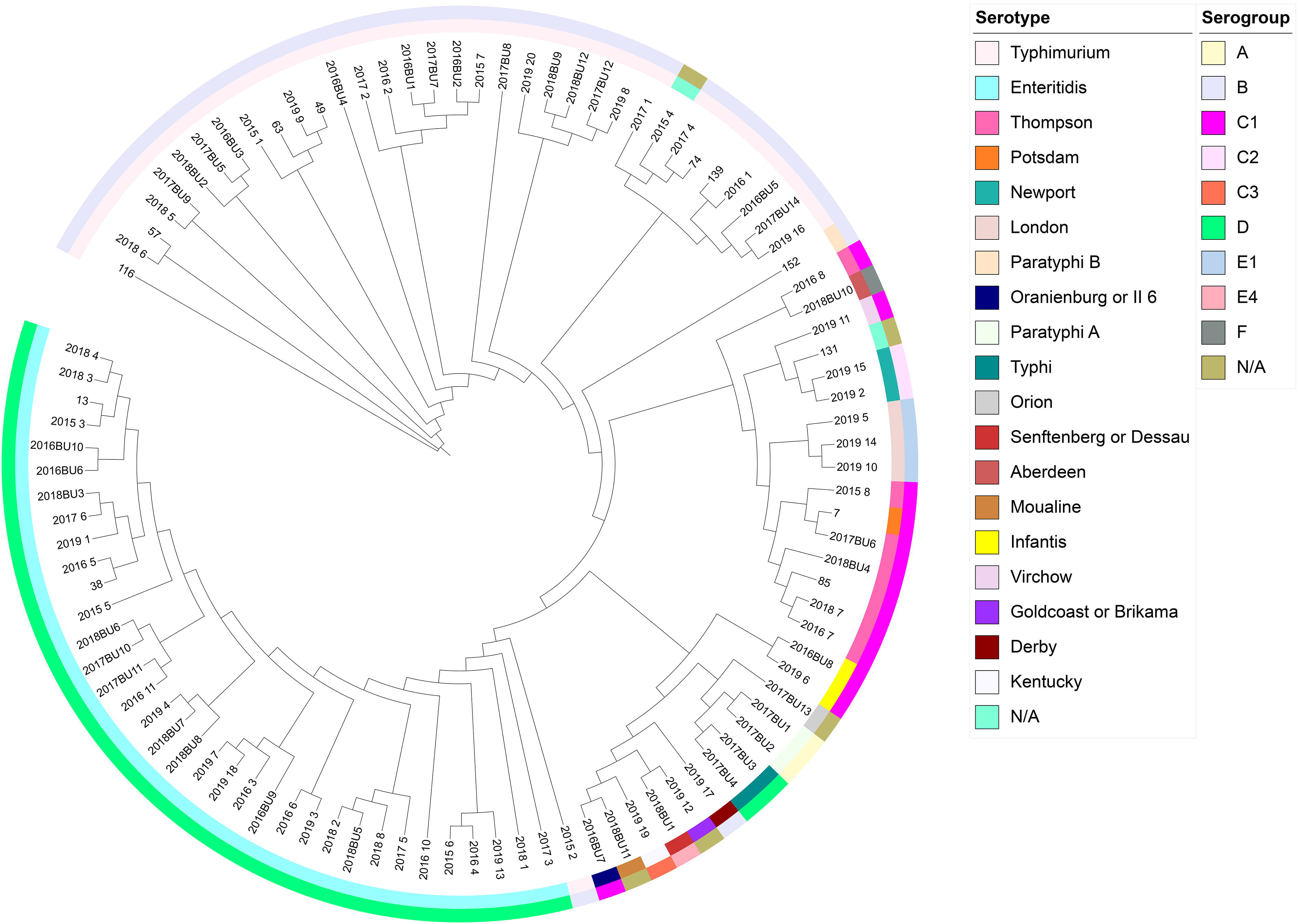
Phylogenetic analysis of Salmonella isolates based on core-genome alignment. The internal layer of the colored circles indicates the serotypes and the external layer of the colored circles indicates the serogroups.
Based on the clinical medical records, the temporal distribution of these Salmonella isolates is shown in Figure 4. We found that the prevalence of Salmonella is periodic, and almost all the Salmonella isolates used in this study were isolated from April to October of the respective year, peaking in August. Figure 5 illustrates an MLST-based minimal spanning tree of all tested isolates. The most prevalent sequence types ST11, ST19, and ST34, appear every year, followed by ST26, which appeared in 4 of the 5 studied years. Interestingly, the other STs appeared in only 1 year, except for ST32.
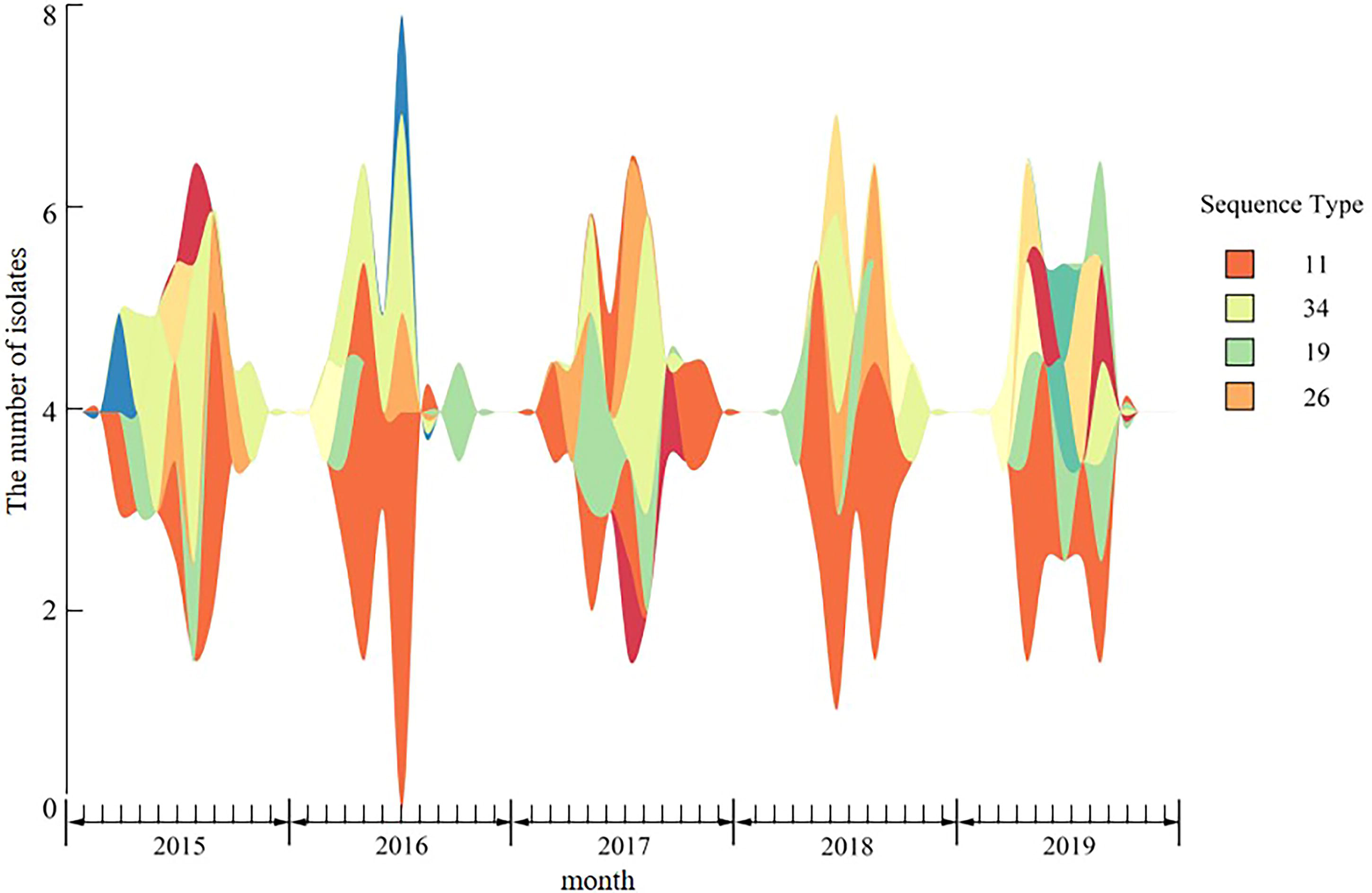
Dynamic prevalence of Salmonella from 2015 to 2019.
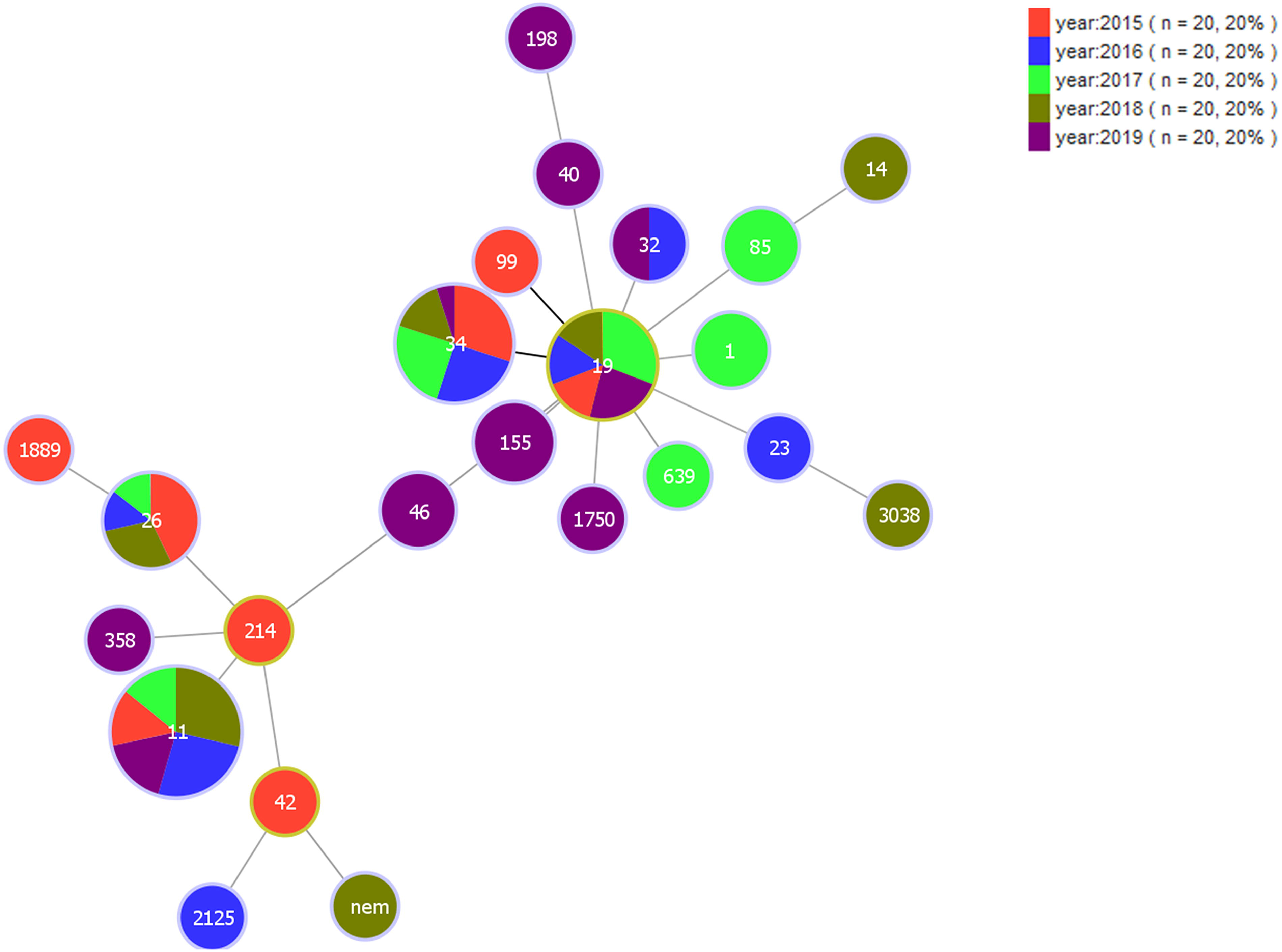
MLST-based minimal spanning tree of Salmonella isolates.
High concordance between serotype and virulence-associated gene profile
With the rapid development of next-generation sequencing, it is easy to acquire genome sequences of bacterial pathogens for scientific research and a huge number of databases have been established based on these sequences. In this study, a VFDB database was used to systematically screen known/potential VFs in the genomes of the 100 Salmonella isolates used in this study.
Several virulence genes/clusters are conserved among these isolates, including the fimbrial adherence determinant gene cluster Agf/Csg, Bcf, Fim, Stb, Std, and Sth, macrophage inducible gene mig-14, magnesium uptake gene cluster Mg2+ transport, and nonfimbrial adherence determinant genes misL, sinH, and PhoPQ regulator. The secretion systems TTSS-1 and TTSS-2 and their translocated effectors were all detected in these Salmonella isolates with different gene composition patterns (Fig. 6).
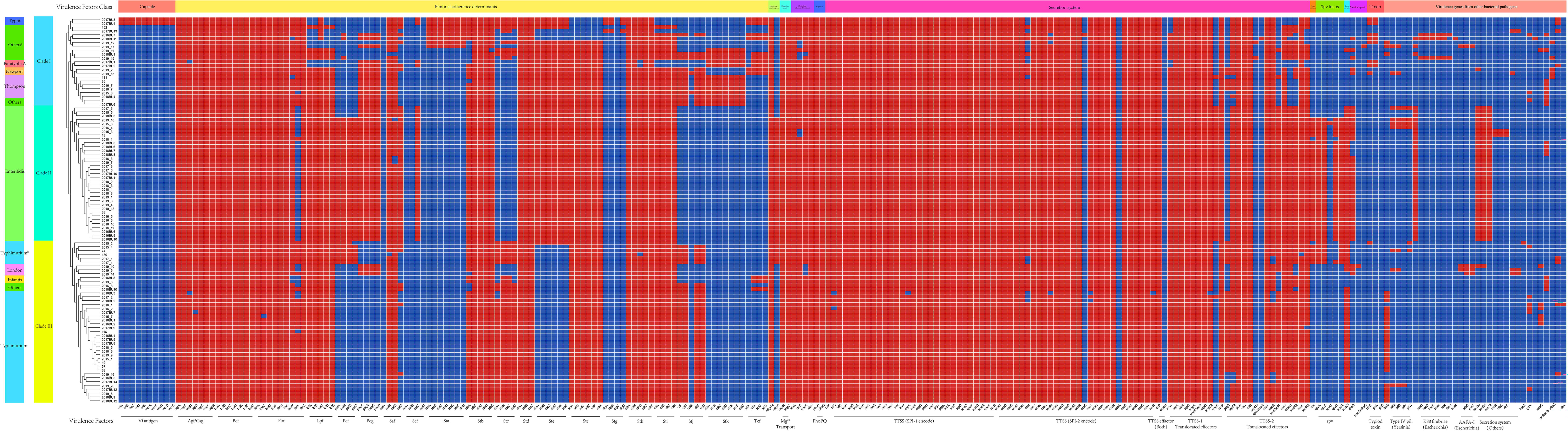
Virulence-associated gene profile of Salmonella isolates. The classes of virulence factors are listed in the color strips above the heatmap. The serotypes of Salmonella are listed in the left color strip.
Based on the presence and absence of VFs, the Salmonella isolates used in this study were clustered into three clades (Fig. 6). S. Enteritidis isolates showed a different virulence gene profile from other serotypes (Clade II), followed by S. Typhimurium isolates which all belong to Clade III. However, it could not be distinguished from S. London, S. Infantis, and two other isolates. Clade I is composed of multiple serotypes. We also observed that there are differences in virulence gene distribution among different serotypes, such as capsule antigen Vi which is only detected in S. Typhi isolates.
In addition, virulence genes in other bacterial pathogens were also screened in this study. Interestingly, we found that the presence of some of these virulence genes is related to serotypes, such as an Escherichia coli brain endothelial cell invasion gene ibeB, which could be detected in almost all Clade I isolates but was absent from Clade II, whereas Escherichia coli ACE T6SS is restricted to Clade II. Our findings demonstrated that the virulence gene profile and horizontal transfer are highly concordant with the serotype.
Association between antimicrobial resistance genotype and phenotype
Almost all these Salmonella isolates were susceptible to imipenem (98/100), followed by azithromycin (90/100), gentamicin (89/100), cefoxitin (88/100), ceftazidime (87/100) and cefotaxime (85/100). However, over half of these collections exhibited resistance to ampicillin (71/100), nalidixic acid (55/100), and tetracycline (52/100). Nearly half (47/100) of Salmonella isolates were determined to be multidrug-resistant (MDR, resistant to ≥3 antimicrobial agents).
An in silico method was exploited to screen the acquired ARGs and chromosomal mutations among these Salmonella isolates. The aac(6’)-Iaa gene was detected in all isolates, followed by sul2 (63/100) and blaTEM-1B (61/100) (Table 1), and at least one QRDR (quinolone resistance-determining region) mutation was detected in 69 isolates (69/100) (Supplementary Table S1). Subsequently, the correlation between the antimicrobial resistance genotype and phenotype was assessed. Interestingly, the presence of ARGs or chromosomal mutations was associated with the antimicrobial resistance phenotype, though the association was not strict. By calculating the positive/negative predictive value, only chloramphenicol and azithromycin achieved over 80% precision in both values. Positive/negative predictive value precision of under 40% was found in three antimicrobial agents: trimethoprim–sulfamethoxazole, imipenem, and ceftazidime (Table 2).
Antimicrobial Resistance Genes of the Salmonella Isolates
The Performance of Whole-Genome Sequencing in Determining the Antimicrobial Susceptibility of Salmonella Isolates
Antimicrobial resistance differs between the major Salmonella STs
The antimicrobial resistance profiles of the four most common STs were compared. There was a significant variation among these major STs in AMR profiles. ST19 and ST26, which are the major sequence types in S. Enteritidis and S. Thompson, showed a higher proportion of resistance to most classes of antibiotics. In contrast, S. Typhimurium ST11 and ST34 exhibited significant resistance to ampicillin and the quinolone class antimicrobial agents, but most of these isolates were resistant to only two or three antimicrobial classes, which was less than that of other isolates (Fig. 7A).
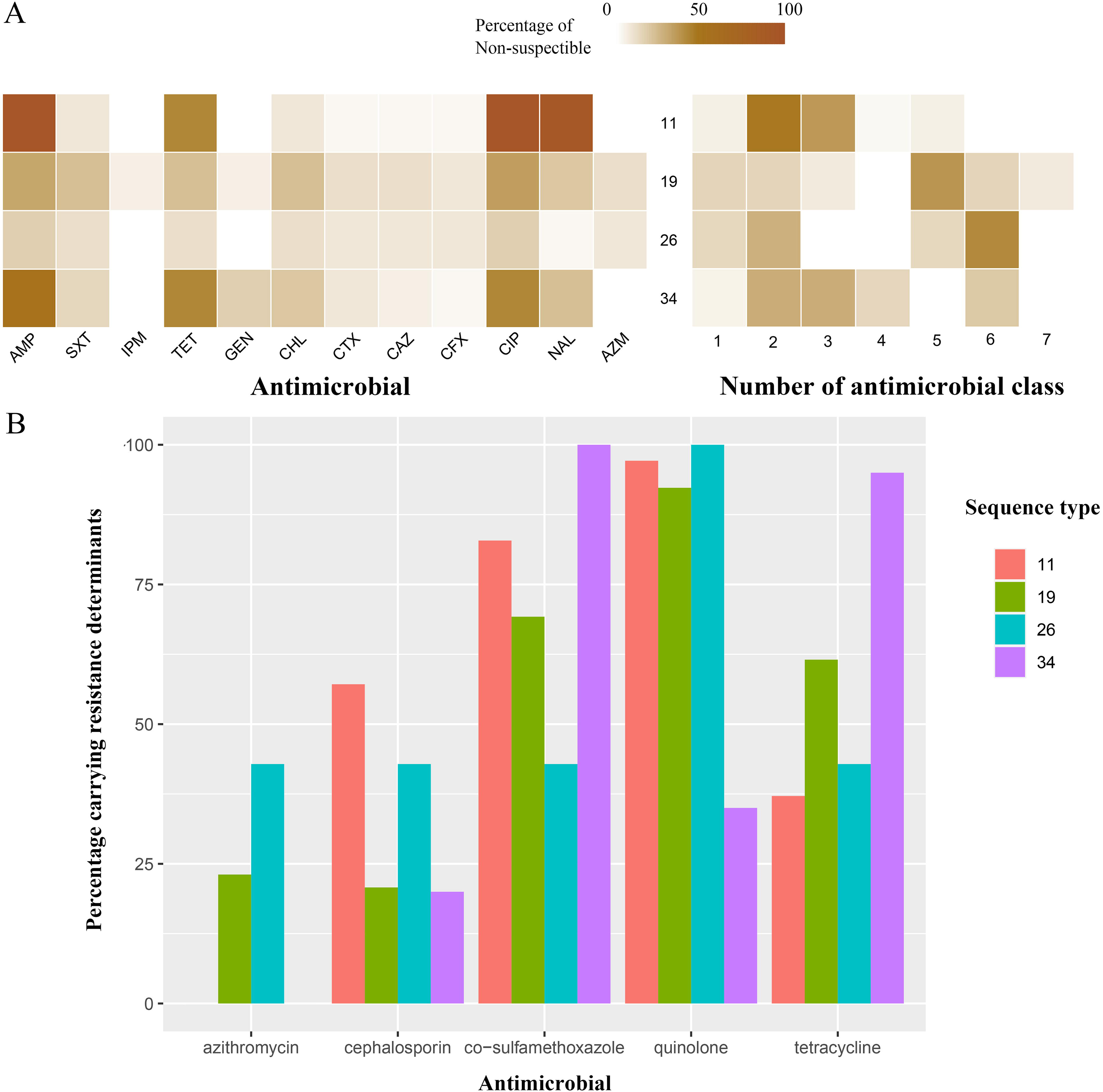
Antimicrobial resistance patterns in major Salmonella sequence types.
The antimicrobial genotype between these sequence types is also different. ST34 exhibited a higher percentage of isolates carrying genes resistant to cosulfamethoxazole (100%) and tetracycline (95%), and the percentage of other isolates was low. ST34 isolates exhibited a significantly lower percentage of isolates carrying genes resistant to quinolones (35%, others >90%). More than half of the ST19 isolates had genes conveying resistance to cosulfamethoxazole, quinolone, and tetracycline, but the percentage of isolates carrying resistance genes to azithromycin and cephalosporin was lower than 25%. The resistance gene profiles of ST11 and ST26 also had their own characteristics (Fig. 7B).
Discussion
The demographic analysis revealed a notable predilection for pediatric salmonellosis in Jiangsu Province, with 56% of isolates originating from male children aged 0–16 years. This aligns with global observations that children under 5 years old are disproportionately affected by salmonellosis due to immature immune systems and inadequate hygiene practices. 20 The elevated proportion of cases in children ≤3 years (44%) further underscores their vulnerability. Our data underscore the need for targeted surveillance and preventive measures in childcare settings. The prevalence of Salmonella has seasonal and cyclical patterns, which is similar to the findings of studies conducted in Thailand, 21 Vietnam, and other provinces of China.22,23 This prevalence period may coincide with the subtropical monsoon climate in this geographical location, as seasonally rainy conditions and humidity could make the transmissibility of bacterial pathogens easier. This indicates that the relevant departments should pay particularly during the rainy season (from April to October), so as to detect the epidemic situation in a timely manner.
The phylogenetic analysis demonstrated tight clustering of isolates within serogroups B and D, reflecting the evolutionary conservation of serotyping markers.24,25 The dominant serotypes are S. Enteritidis and S. Typhimurium, which is similar to a separate series of Salmonella surveillance studies in China.26–28 Notably, S. Enteritidis exhibited clonal homogeneity (ST11 only), while S. Typhimurium displayed genetic diversity (4 STs), which is similar to Yan’s report. 29 The most prominent ST in this study was ST11, which differs from prior reports in which ST34, which is strongly associated with food production, was the dominant sequence type. 30 ST19 and ST26 have also been frequently reported in Asia.31,32
The conserved virulence genes (e.g., Agf/Csg, TTSS-1/2) across isolates highlight their essential roles in pathogenesis. However, serotype-specific differences were striking: Vi antigen was exclusively detected in S. Typhimurium, consistent with its role in evading host immunity. 33 Cross-species virulence genes (e.g., ibeB from E. coli) exhibited clade-specific distribution, and the horizontal transfer of these genes suggests adaptative evolution through interspecies recombination—a mechanism previously implicated in Salmonella virulence diversification. 34 This concordance between serotype and virulence profile has clinical implications for diagnostic marker development.
The high rates of ampicillin, tetracycline, and quinolone resistance underscore widespread plasmid-mediated resistance (e.g., blaTEM-1B, tetA) and chromosomal mutations (e.g., QRDR mutations).
Notably, 47% of isolates exhibited MDR, posing significant treatment challenges. The dominance of ARGs like aac(6′)-Iaa and sul2 reflects plasmid-mediated resistance mechanisms, common in Asia. However, we found that the association between antimicrobial resistance genotype and phenotype is not strict. These findings differ from those of studies in other countries, such as Vietnam and the United Kingdom, 22 and there may be some differences in the genomic evolution of Salmonella isolates from different geographical locations, leading to a different phenotype.
Comparative analysis of major STs revealed divergent resistance patterns. S. Enteritidis ST19 and S. Thompson ST26 exhibited broad resistance to multiple classes, contrasting with S. Typhimurium ST11/ST34, which showed selective resistance to β-lactams and quinolones. These findings emphasize the need for ST-guided antibiotic stewardship to optimize treatment efficacy and curb resistance propagation.
Salmonella is one of the most common bacterial pathogens clinically. In this study, we observed that children under 3 years old were the most susceptible group, the serotypes S. Typhimurium and S. Enteritidis were the most prevalent serotypes and severe multidrug resistance was observed in this study. Our work also shows the association between serotype, antimicrobial resistance, and virulence gene profile and demonstrates the connection between genotype and phenotype, providing epidemiological data on Salmonella.
Footnotes
Acknowledgment
The authors would like to thank the sentinel hospitals in Jiangsu Province for collecting, isolating, and sending strains.
Authors’ Contributions
X.Q. conceptualized and edited the article. H.C. wrote the article and prepared the figures. K.M. was involved in data curation. D.Z. was involved in resources and supervision.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.