
Abstract

However, some investigators have suggested that the technique of “Mendelian randomization”—the application of instrumental variables to genetic epidemiology—provides a shortcut for this process that can readily “rule in” or “rule out” the role of putative intermediate phenotypes as “causal” agents for disease. Whether this presumption is true for highly complex disorders like atherothrombosis is uncertain, and the limitations of Mendelian randomization studies are often minimized in the clinical literature. Null Mendelian randomization studies that purport to “rule out” a causal pathway are particularly problematic to interpret. First, the proportion of variance explained by genetic instrumental variables in studies of atherosclerosis has typically been very small, an effect greatly limiting all studies other than those with exceptional sample size. Second, it has often been impossible to verify that the genetic effect is independent of all other pathways for disease except that mediated through the biomarker of interest. Third, a null Mendelian randomization data for a biomarker of a given pathway neither addresses nor rules out a causal role for the pathway itself. Heritable epigenetic changes in gene expression that occur without altering the DNA sequence also prove a considerable challenge to the underlying precepts of Mendelian randomization.
Finally, when considered from a clinical perspective, whether or not a biomarker is “causal” should have little if any influence on decisions regarding its role in daily practice; temperature is a crucial biomarker of infection that physicians do not ignore simply because it is a result rather than a cause of pneumonia. Better understanding of both the merits and limitations of Mendelian randomization will improve interpretation of these studies.
How Does Mendelian Randomization Begin to Address Causal Relationships?
Mendelian randomization is an application to genetics and medicine of instrumental variables as a way of inferring causality in nonexperimental settings. Initially developed by economists to better understand policy shifts in econometric models, an instrumental variable is a variable that is substantially associated with the outcome of interest only through its association with the exposure or biomarker of interest such that there are no other alternative pathways that might link the instrumental variable to the outcome. 1 In 1986, Katan made the first application of this approach to medicine, suggesting that genetic variants could serve as an instrumental variable and that they might be useful to reduce confounding and help to establish causal relationships between an intermediate phenotype and disease. 2,3 Because alleles are randomly allocated at the time of gamete formation, the use of polymorphism data as instrument variables would later come to be known as “Mendelian randomization.” 4
Figure 1a illustrates this approach where biologic interest lies in the potential “causal” relationship of a biomarker B [such as low-density lipoprotein (LDL) or glycosylated hemoglobin (HbA1c)] with subsequent occurrence of a disease D (such as myocardial infarction or diabetes), yet ascertainment of this relationship is difficult due to the presence of multiple potential confounders C (such as smoking or obesity). In this situation, if there is a polymorphism at a genetic loci G that in turn is strongly associated with B and unrelated to C, then the finding of a robust association between G and D can be used to make an argument for causality between B and D. Assuming a random distribution of alleles, this use of genetic information as instrumental variables has the potential to reduce or avoid some of the limitations of observational epidemiology, in particular reverse causality, confounding, and regression-dilution bias. 5 –7
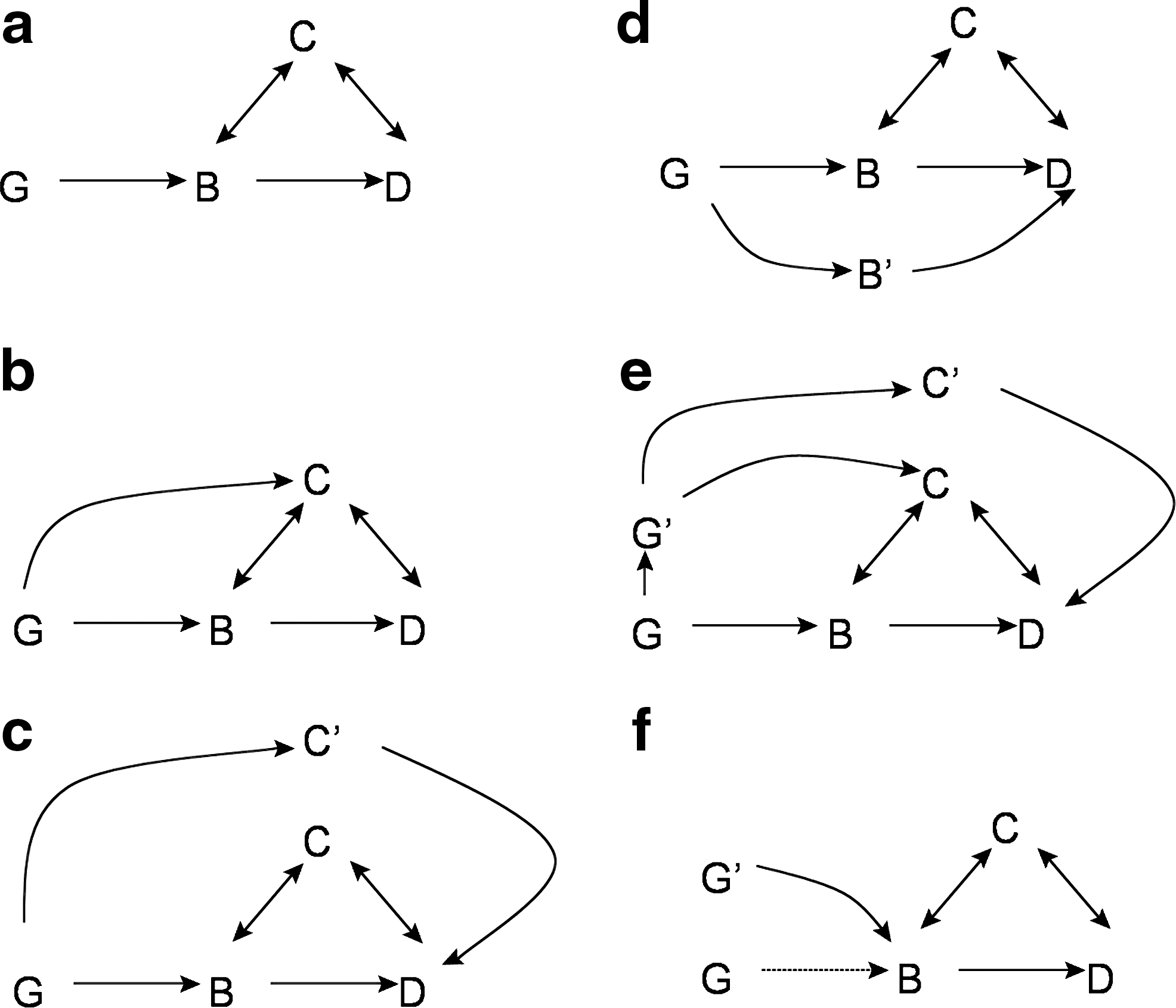
Using genetic data (G) as an instrumental variable, Mendelian randomization can help to address whether a biomarker (B) is causally related to disease (D) as long as the relationship of G to D is only through B and not through any other pathway. In such situations, a known confounder (C) of the relationship between B and D should not result in a false-positive finding (
The minimal conditions for such an interpretation to be made are: (1) The instrument variable G has substantial impact on B; (2) that G affects the outcome D only through B and that there is no other pathway or intermediate phenotype linking G to D; and (3) that there is no direct or indirect confounding of the effect of G on D. An example of this kind of association would be of a randomly assorted locus distributed during meiosis that is strongly associated with LDL cholesterol (LDL-C) and that is known to impact upon myocardial infarction through no possible alternative pathway, except through its influence on LDL-C. In such a setting, if increased event rates are observed with alleles that mark LDL-C, we can with reasonable confidence suggest a “causal” relation between LDL-C and myocardial infarction. Thus, at least in theory, Mendelian randomization studies can provide a valuable and novel method to make inferences about causal relationships within the context of observational cardiovascular epidemiology.
What Are the Limitations of Mendelian Randomization?
As many authors have recently described, 5 –9 the application of Mendelian randomization to a complex disease such as atherothrombosis with multiple envioronmental, lifestyle, and genetic influences has proven difficult. Table 1 provides a formal listing of limitations that apply to Mendelian randomization in general. Figure 1, b–f, outlines multiple genetic epidemiology situations that can render Mendelian randomization studies ineffective.
Among these practical limitations, several have proven particularly problematic for cardiovascular epidemiology. First, in many instances, the critical assumption that G influences D only through B has either been unverifiable or frankly contradicted. Rather, our understanding of the genetics of heart disease has instead suggested considerable pleiotropy because single genes have turned out to influence a variety of intermediate phenotypes and thus other pathways beyond B alone.
Second, other alleles (G′) may correlate with G through linkage disequilibrium and thus lead to an influence on D through secondary pathways, thus violating the concept of a lack of confounding due to “randomization.”
Third, even in studies where known sources of confounding (C) are well ascertained, unknown sources (C′) remain that can provide direct linkage from G to D, or through linkage disequilibrium, from G′ to D.
Fourth, in most instances to date, the proportion of variance in B explained by G (i.e., the magnitude of association) has not been “substantial” but in fact has been very small. This is particularly problematic when interest focuses on estimation of odds ratios or relative hazards in multiplicative models. Moreover, when G explains little of the variation in B, a Mendelian randomization study offers only marginal insight into the relevance of an underlying pathway that B may mark. As a consequence, distinguishing informative null Mendelian randomization studies from those that simply have inadequate power or inadequate representation of the pathway is difficult.
Finally, the application of instrumental variables to genetic epidemiology has been hampered by the fact that the level of genetic heterogeneity underlying cardiovascular disease is uncertain; that the role of gene–gene and gene–environment interactions is likely to be substantial; that effects due to canalization, developmental compensation, population stratification, linkage disequilibrium, and epigenetics have been unanticipated; and that the availability of identifying functional variants has to date been limited. 5,6,9
How Should We Interpret Causal Relationships in Both “Positive” and “Negative” Mendelian Randomization Studies?
Given the above limitations, clinicians and researchers must exercise appropriate caution in interpreting both “positive” and “negative” Mendelian randomization studies.
If verifiable in multiple large-scale prospective cohort settings, replicated findings in “positive” Mendelian randomization studies that G significantly associates with D only through B should increase our enthusiasm that B is a potential causal agent. However, even when these conditions exist, clinicians and researchers must recognize that Mendelian randomization data alone are not sufficient to establish a causal pathway. For example, early Mendelian randomization studies of homocysteine suggested a “causal” relationship, 10 yet multiple trials of homocysteine reduction have since failed to show any benefit on cardiovascular event reduction.
The interpretation of “negative” Mendelian randomization studies provides even greater complexity. While authors of null Mendelian randomization studies may be tempted to claim “these data demonstrate that B has no causal role in disease D,” such a conclusion is rarely if ever justifiable given the high potential for false-negative findings. Even if produced from very large data sets and in settings where the magnitude of effect of G on B is large and where exceptional steps have been taken to reduce the potential for confounding of the relationships between G and D, such studies only reduce but do not eliminate the potential for causality. That process, as in all of science, depends on the totality of evidence from multiple sources, of which Mendelian randomization is only one. If a polymorphism were found to influence smoking behavior but not impact upon subsequent rates of lung cancer or myocardial infarction, would we be comfortable excluding a role for smoking in either of these disorders?
A particularly complex situation arises when both “positive” and “negative” findings appear in the same study population. In our own evaluations of polymorphisms within the CETP gene as determinants of both high-density lipoprotein cholesterol (HDL-C) and vascular risk, we found several single nucleotide polymorphisms (SNPs) that would appear to satisfy most tenets of Mendelian randomization. 11 However, in the same database, we also found other SNPs in the CETP region that were not associated with vascular events. Such simultaneous findings suggest that relationships between genes, intermediate phenotypes, clinical outcomes are typically far more complex than the simple model posed in Fig. 1a.
What Are the Implications of Mendelian Randomization for Biomarkers in Clinical Practice?
An additional source of confusion in the clinical literature has been incorrect use of Mendelian randomization data to “validate” certain biomarkers for clinical use and to “exclude” others. Such confusion arises from the often forgotten fact that clinically useful biomarkers need not have a causal relation to disease. As a simple example, temperature is a crucial biomarker of infection that physicians do not ignore simply because it is a result rather than a cause of pneumonia.
In fact, “causality” per se plays little role in the formal assessment of biomarkers as potentially useful clinical tools. As described in a recent overview from the American Heart Association, 12 risk markers being considered for clinical use should at a minimum: (1) demonstrate consistent independence of effect in multiple prospective cohort studies; (2) demonstrate incremental information on utility beyond that of usual risk factors; (3) demonstrate that assessment leads to clinical impact on patient management and outcomes; and (4) be readily assessed with standardized assays. If “causality” were a criterion for biomarker selection, then use of HbA1c in diabetes or imaging in atherosclerosis would have to be eliminated from clinical practice because these biomarkers, like temperature, are a result of disease rather than a cause.
The clinical confusion that can result from overinterpretation of Mendelian randomization studies is illustrated in ongoing controversy surrounding the role of inflammation in heart disease and the potential clinical role of inflammatory biomarkers such as C-reactive protein (CRP). To date, abundant epidemiologic, pathophysiologic, and experimental data support a crucial role for inflammation in all phases of atherothrombosis. 13 Furthermore, for the inflammatory biomarker high-sensitivity CRP (hsCRP), more than 50 prospective cohort studies worldwide consistently indicate that levels of hsCRP linearly predict future cardiovascular risk with a magnitude of effect larger than that of total or non-HDL-C. 14 Following the observation that statin therapy lowers hsCRP in a manner largely unrelated to LDL-C reduction, 15,16 randomized clinical trials were performed indicating that individuals with average to low levels of cholesterol who are nonetheless at increased vascular risk due to increased hsCRP levels markedly benefit from statin therapy 17,18 and that the magnitude of this benefit is related not only to the levels of LDL-C achieved, but also to the levels of hsCRP achieved. 19 –22
It is on the basis of this totality of evidence that organizations including the American College of Cardiology, the American Heart Association, the National Academy of Clinical Biochemistry, the United States Food and Drug Administration, and the Canadian Cardiovascular Society have endorsed the use of hsCRP as a method to identify populations at increased cardiovascular risk that will benefit from lifestyle interventions and statin therapy. 23 –26 Within this context, findings from Mendelian randomization studies that any one inflammatory biomarker does or does not play a “causal” role in atherothrombosis would have little relevance to clinical practice. Equally important, even if a definitive Mendelian randomization study was performed for any single inflammatory biomarker, such a demonstration would still say little about the broader role of inflammation as a potential causal process for atherothrombosis. Only rigorous experimental science performed at the bench and innovative randomized trials of targeted antiinflammatory agents will be able to address these latter questions.
Footnotes
Author Disclosure Statement
Dr. Ridker is listed as a co-inventor on patents held by the Brigham and Women's Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease, and has received research support for genetic epidemiology from the National Heart Lung and Blood Institute, Amgen, Celera, and Roche Diagnostics. Dr. Ridker was the Principal Investigator and Dr. Glynn the academic study statistician for the JUPITER trial which was funded by AstraZeneca. Drs. Danik-Suk and Paynter report no conflicts relevant to this article.