
Abstract
Bile acids are not only intestinal detergents, but also act as signaling molecules to control bile acid and metabolic homeostasis. By binding to the nuclear receptor farnesoid X receptor (FXR) or the membrane receptor TGR5, bile acids regulate lipid, glucose, and energy metabolism. The removal of bile acids from the enterohepatic cycle by orally administered sequestrants, nonabsorbable resins that complex bile acids in the intestinal lumen, is an effective treatment to reduce plasma cholesterol concentrations in dyslipidemia. In diabetic patients, sequestrants further lower hyperglycemia, and are thus useful in glycemic control. Here, we review the signaling pathways involved in the glucose-regulatory function of bile acids and our current understanding of the mechanisms behind the glucose-lowering effect of bile acid sequestrants.
Introduction
In addition to their function in lipid digestion, bile acids are increasingly studied for their metabolic regulator roles. By triggering different signaling pathways, bile acids participate in the control of bile acid, lipid, and glucose homeostasis. Interestingly, modulation of bile acid metabolism by intestinal sequestration leads to improved glycemic control in T2DM patients, indicating a strong link between bile acid and glucose homeostasis. In this review, we summarize the signaling pathways through which bile acids can exert control on glucose homeostasis. We further discuss potential mechanisms explaining the glucose-lowering effect of bile acid sequestrants (BAS) by integrating the available evidence from animal and clinical studies.
Bile Acids and the Enterohepatic Cycle
Bile acids are amphiphatic molecules synthesized from cholesterol in the liver and stored in the gallbladder between meals. Upon ingestion of a meal, bile acids are released into the intestinal lumen, where they form micelles with dietary lipids and lipid-soluble vitamins, facilitating their absorption. Only 5% of bile acids are excreted in the feces. The majority are reabsorbed in the distal ileum and transported back to the liver, thus completing the enterohepatic cycle.
The human liver produces the primary bile acids CA and chenodeoxycholic acid (CDCA) and their glycine and taurine conjugates. The key enzymes of bile acid synthesis are CYP7A1 and sterol-12α-hydroxylase (CYP8B1). CYP7A1 determines the size of the bile acid pool whereas CYP8B1 defines the ratio of CA to CDCA and thus the pool composition and hydrophobicity. Once primary bile acids have reached the intestine, they may undergo deconjugation and dehydroxylation by bacteria of the microflora, giving rise to secondary bile acids, of which the most important are DCA and lithocholic acid (LCA) in humans. After completion of another enterohepatic cycle, a small part of the secondary bile acids is again modified in the liver to form tertiary bile acids. These processes are finely regulated, e.g. by bile acids themselves, to assure the maintenance of a bile acid pool of defined size and composition. 8
BAS as Glucose-Lowering Agents
Bile acid metabolism can be pharmacologically modulated by sequestrants (cholestyramine, colesevelam, colestimide, colestipol), nonabsorbable resins that bind bile acids in the intestinal lumen. The formed complex is secreted in the feces and thus diverts bile acids from the enterohepatic cycle. 9 To maintain the bile acid pool, bile acid synthesis is upregulated and the delivery of low-density lipoproteins (LDL) to the liver is enhanced to provide cholesterol as a substrate. As a consequence, plasma LDL levels decrease. 10 BAS have been used for over 25 years for the therapy of dyslipidemia, and several clinical studies have indicated the benefit of their cholesterol-lowering effect for the prevention of coronary heart disease (reviewed in detail in refs. 9 and 11).
In addition to its lipid-lowering effect, the sequestrant colesevelam was approved in 2008 as an adjunct to diet and exercise to improve glycemic control in adults with T2DM. Colesevelam HCl has not been approved for use for glycemic control in type 1 diabetes or for treating diabetic ketoacidosis and has not been studied in T2DM as monotherapy or in combination with dipeptidyl peptidase 4 inhibitors, or extensively with thiazolidinediones. The first observation for such an effect of BAS came from a randomized, double-blind, crossover study in which cholestyramine was administered to T2DM patients for 6 weeks. The treatment improved glycemic control, as evidenced by a reduction of plasma glucose levels, a decrease in urinary glucose excretion, and a tendency to lower glycosylated hemoglobin (HbA1c) levels. 12 Subsequent studies confirmed this beneficial effect of BAS in T2DM patients. Zieve et al. measured reduced HbA1c, plasma fructosamine, and postprandial glucose levels after 12 weeks of colesevelam HCl treatment, 13 and Yamagata et al. reported that 12 weeks of colestimide treatment decreased HbA1c and fasting plasma glucose concentrations. 14 Furthermore, colesevelam HCl improved glycemic control in patients with T2DM who were not adequately controlled by insulin, sulfonylurea, or metformin therapy. 15 –17 These clinical data suggest a link between bile acid and glucose metabolism. Whether the modulation of bile acid metabolism also positively affects glycemic control in T1DM patients has not yet been addressed.
Bile Acids as Regulators of Glucose Homeostasis
Over the past decade, evidence has accumulated identifying bile acids as mediators of metabolic control. Bile acids exert differential regulatory functions via nuclear and membrane receptors as well as via receptor-independent signaling pathways. Some of these pathways may participate in the control of glucose metabolism by bile acids.
Nuclear receptors: FXR signaling
Bile acids activate nuclear receptors, transcription factors regulating expression of target genes by binding to specific response elements in their promoter. Hepatocyte protection from bile acid cytotoxicity occurs via LCA activation of the pregnane X receptor (PXR), the vitamin D receptor (VDR), and the constitutive androstane receptor (CAR), which subsequently induce genes involved in bile acid detoxification and elimination. 18 However, the best-characterized nuclear receptor mediating the regulatory actions of bile acids in bile acid, lipid, and glucose metabolism is the farnesoid X receptor (FXR). 19 FXR displays high expression levels in liver and intestine and lower expression in adipose tissue, pancreas and the adrenal glands. FXR is most effectively activated by CDCA. 8 Two distinct mechanisms have been described by which FXR mediates bile acid signaling. In the liver, FXR activation induces the expression of the orphan nuclear receptor short heterodimer protein (SHP), which subsequently inhibits a number of transcription signal transduction pathways—the liver receptor homolog 1 (LRH1) induction of CYP7A1, hepatocyte nuclear factor 4α (HNF4α) activation of gluconeogenic genes, i.e., phosphoenol pyruvate carboxykinase (PEPCK), and sterol regulatory binding element 1c (SREBP1c) induction of lipogenesis. The second mechanism implies the induction of the fibroblast growth factor 19 (FGF19, FGF15 is the murine homolog) by FXR in the intestine. Circulating FGF19 binds to the FGF receptor 4 (FGFR4) in the liver, thus activating the c-Jun N-terminal kinase (JNK) pathway, which blocks the HNF4α-mediated activation of CYP7A1.
The first observation that linked FXR, and thus bile acids, to glucose metabolism was the fact that hepatic FXR expression is dysregulated in rodent models of T1DM and T2DM. Mechanistic studies in rat hepatocytes have demonstrated that FXR expression is repressed by insulin and that this effect is reversed in the presence of glucose. 20 FXR-deficient mice revealed a role for FXR in the regulation of the dynamics of glucose metabolism. The induction of glycolytic [liver pyruvate kinase (LPK)] and lipogenic genes [fatty acid synthase (FAS) and acetyl CoA carboxylase (ACC)] is accelerated during the transition from fasting to high-carbohydrate refeeding in the absence of FXR, associated with decreased hepatic glycogen stores and a transient hypoglycemia. Conversely, FXR activation attenuated the induction of glucose-responsive genes, possibly due to the interference of FXR with the carbohydrate response element binding protein (ChREBP), an activator of glucose-induced LPK, FAS, and ACC expression. 21,22 Finally, FXR-deficiency also results in altered kinetics of intestinal glucose absorption. 23
In addition to the regulation of glucose utilization, bile acids impact hepatic glucose production. FXR-deficient mice displayed a decreased expression of the gluconeogenic genes PEPCK 21,22,24 and glucose-6-phosphatase (G6Pase), 24 suggesting that FXR positively regulates gluconeogenic gene expression. The induction of PEPCK expression in mice by FXR activation with the synthetic agonist GW4064 confirmed this finding. 25 However, FXR activation by dietary CA decreased PEPCK and G6Pase 24 as well as fructose-1,6-bisphosphatase (FBP1) expression 26 in a FXR- and SHP-dependent manner. Furthermore, a FXR-independent mechanism for the negative regulation of PEPCK by bile acids was proposed via the repression of HNF4α-mediated activation of PEPCK expression. 27 Even though the precise regulations and mechanisms of gluconeogenic gene expression by bile acids are unclear, it appears that bile acids may also affect hepatic glucose production.
In addition to its function in hepatic glucose metabolism, FXR has been identified as a modulator of insulin sensitivity. FXR-deficient mice display insulin resistance with a reduction of peripheral glucose disposal and decreased insulin signaling in adipose tissue and skeletal muscle. 24,28,29 Because FXR is not expressed in skeletal muscle, the impaired insulin sensitivity in this tissue was attributed to a secondary effect exerted by elevated circulating fatty acids. 24 Whether FXR-deficient mice also exhibit hepatic insulin resistance is unclear. 22,24,28 Further studies in liver or adipose tissue-specific FXR-deficient animals are necessary to resolve this issue. Pharmacological activation of FXR in diabetic rodent models attenuated insulin resistance. GW4064 treatment of leptin-deficient (ob/ob) mice reduced hyperinsulinemia and improved glucose tolerance. 29 The semisynthetic FXR agonist 6-ethyl-CDCA reversed insulin resistance with improved liver and skeletal muscle insulin sensitivity in leptin receptor-deficient ( fa/fa) rats. 30
Changes in energy metabolism may lead to a modulation of glucose homeostasis. FXR is expressed in adipose tissue, where it controls adipocyte differentiation and function. 29,31 FXR-deficient mice have a reduced adipose tissue mass associated with a disturbed glucose homeostasis. 24,28,29 It would be interesting to evaluate the potential changes in adipose tissue biology and the consequences on energy and glucose homeostasis in models of obesity and diabetes. FGF19, which is induced by intestinal FXR, affects energy and glucose metabolism. Administration of human FGF19 reversed weight gain and insulin resistance in mouse models of genetic and diet-induced obesity, due to an increase in metabolic rate and hepatic fatty acid oxidation. 32 The concept of metabolic regulation by bile acids via the intestinal FXR–FGF19 axis is appealing, but requires more detailed investigations.
Membrane receptors: TRG5 signaling
TGR5 (also known as Gpbar1, M-Bar, and BG37) is a G protein-coupled membrane receptor activated by certain bile acids. 33,34 Mainly expressed in gallbladder, ileum, colon, brown and white adipose tissue, and, to a lesser extent, in skeletal muscle and liver, TGR5 is activated by nanomolar concentrations of LCA and taurine-conjugated lithocholic acid (TLCA) and micromolar concentrations of CA, DCA, and CDCA. 35 Upon ligand binding to TGR5, the GαS subunit is released and activates adenylate cyclase. The resulting increase in cyclic adenosine monophosphate (cAMP) concentrations leads to a consecutive activation of protein kinase A (PKA) and the transcription factor cAMP response element-binding protein (CREB). 36 TGR5 can also activate the mitogen-activated protein kinase (MAPK) pathway. 18
Even though TGR5-deficient mice displayed no changes in glucose metabolism, 37,38 bile acids can exert TGR5-mediated control on glucose homeostasis by affecting the incretin system. Incretins such as glucagon-like peptide 1 (GLP1) and glucose-dependent insulinotropic peptide (GIP) are intestinal hormones, secreted upon ingestion of food, that stimulate pancreatic insulin secretion. LCA and DCA induce GLP1 secretion from intestinal L cells in vitro. 39 TGR5-overexpressing mice fed a high-fat diet did not develop glucose intolerance and had elevated GLP1 levels leading to an increased insulin release in response to an oral glucose load. The proposed mechanism involves a TGR5-mediated increase of the adenosine triphosphate (ATP) to adenosine diphosphate (ADP) ratio that subsequently leads to increased intracellular calcium concentrations that trigger GLP1 secretion. Pharmacological activation of TGR5 by the semisynthetic TGR5 agonist 6-ethyl-methyl CA 40 or by oleanolic acid 41 also improved diet-induced obesity and insulin resistance.
Bile acids have been attributed a regulatory function in energy metabolism that is mediated by TGR5. CA supplementation to a high-fat diet increased energy expenditure in mice and subsequently alleviated obesity and insulin resistance. 42 The alteration of energy expenditure was linked to a TGR5-mediated increase in cAMP levels, resulting in an enhanced activation of deiodinase (D2), an enzyme that intracellularly converts the thyroid hormone thyroxine (T4) into active triiodothyronine (T3). However, TGR5-deficient mice do not exhibit a difference in weight gain, and only female mice have a higher susceptibility to diet-induced obesity. 37 TGR5-overexpressing mice fed a high-fat diet did not display a difference in weight gain either. Thus, the importance of the TGR5–D2 axis for the regulation of body weight remains elusive. However, a recent study found that thyroid hormones exert a central effect by inhibiting hypothalamic adenosine monophosphate-activated protein kinase (AMPK), subsequently enhancing sympathetic activity, which evokes changes in parameters of energy metabolism in brown adipose tissue. 43 Thus, both peripheral and central actions may play a role in the effects of thyroid hormones on energy homeostasis.
Receptor-independent signaling
Bile acids also activate signaling networks such as the MAPK pathway or induce intracellular calcium mobilization in a receptor-independent manner (extensively reviewed in ref. 18). The contribution of these pathways to metabolic control is however currently unknown. Interestingly, taurine-conjugated ursodeoxycholic acid (TUDCA) has been reported to attenuate insulin resistance and hepatic steatosis in mouse models of obesity and diabetes in a receptor-independent manner. Functioning as a chemical chaperone, TUDCA attenuates endoplasmatic reticulum (ER) stress in vitro and in vivo, a phenomenon implicated in the development of diabetes. 44
Potential Glucose-Lowering Mechanisms of BAS
Sequestration of bile acids improves glycemic control in T2DM patients. 11,13,15 –17 As a direct consequence of their activity, sequestrants alter the composition of the bile acid pool. Colesevelam treatment of T2DM patients resulted in a more hydrophilic bile acid pool due to an increase of CA synthesis. 4 Such changes in pool composition may result in subtle changes of the activity of bile acid-regulated pathways, including FXR and TGR5, which may contribute to the overall improvement in glucose homeostasis. Such effects are, unfortunately, difficult to delineate. Mechanistic data from animal and clinical studies are still scarce. Nevertheless, a growing body of data on bile acid-mediated regulation of glucose homeostasis provides numerous indications for potential mechanisms (Fig. 1).
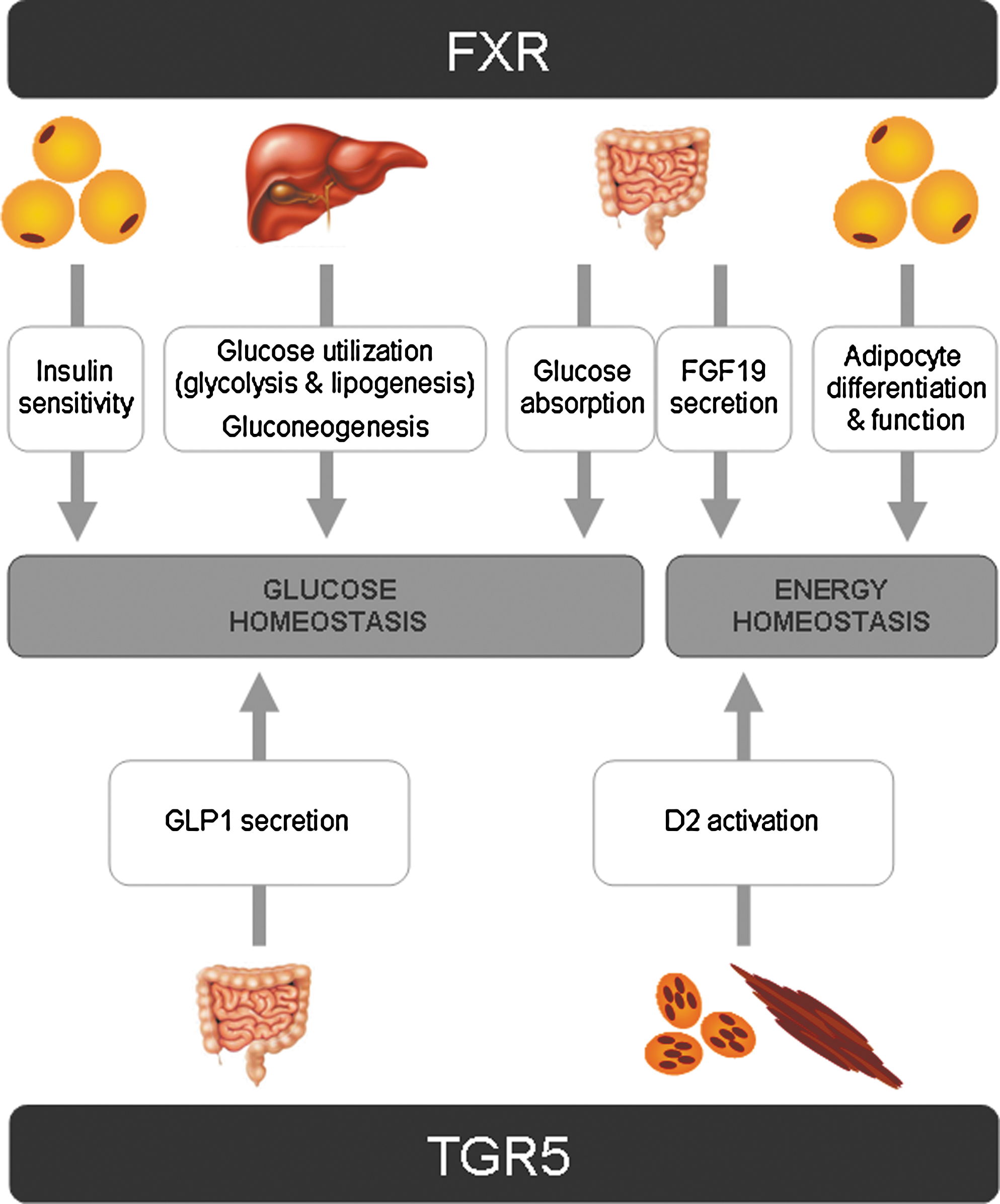
Farnesoid X receptor (FXR)- and TGR5-mediated regulation of glucose and energy homeostasis by bile acids. See text for details. FGF 19, fibroblast growth factor 19; GLP1, glucagon-like peptide 1; D2, deiodinase.
First, BAS may influence intestinal function. An early study suggested a reduction of glucose absorption by sequestrants. 45 These data are in line with observations in FXR-deficient mice, which show a delay in glucose absorption. 23 The TGR5-mediated increase of GLP1 secretion by intestinal L cells 39,40 presents a powerful mechanism by which changes in bile acid metabolism can modulate glucose homeostasis. Recently, two studies have reported that the administration of cholestyramine and colesevelam improved insulin resistance in diabetic rat models by increasing GLP1 release. Both studies exclude a participation of the FXR pathway, because FXR activity was reduced in liver and intestine. 46,47 In humans, colestimide treatment for 1 week lowered postprandial plasma glucose levels in T2DM patients and was associated with elevated postprandial GLP1 concentrations. 48 A more recent study reported that colesevelam treatment for 12 weeks improved glucose homeostasis by increasing GLP1 as well as GIP levels in T2D patients, 49 confirming a potential contribution of the incretin system to the beneficial effect of sequestrants on glycemic control.
Second, BAS may modulate hepatic glucose metabolism. In T2DM, hepatic glucose metabolism is strongly dysregulated, i.e., the impaired repression of endogenous glucose production by insulin contributes to hyperglycemia. Because bile acids exert FXR-dependent control on the expression of genes controlling hepatic glucose handling, 22,24 –26 it is conceivable that the sequestrant-mediated changes in FXR activity may affect hepatic glucose production. Further studies are warranted to delineate the contribution of this pathway to the glucose-lowering effect of sequestrants.
Third, BAS may influence insulin sensitivity, which can underlie the improvement of glucose homeostasis. Treatment of spontaneously diabetic mice with colestimide resulted in an enhanced insulin sensitivity and a subsequent decrease of hyperglycemia. 50 In a cohort of T2DM patients, the administration of colesevelam increased whole-body insulin sensitivity, as calculated from glucose and insulin levels during a postmeal tolerance test. 51 However, so far, the underlying molecular mechanisms have not been addressed. Because bile acids can control insulin sensitivity by FXR- as well as TGR5-dependent mechanisms, 24,28,29,40,41 the contribution of these two pathways to a sequestrant-mediated improvement of insulin sensitivity and glucose homeostasis are an interesting area of research.
Finally, improvements of insulin sensitivity and glucose homeostasis can be secondary to changes in energy metabolism. Bile acids may influence energy expenditure in mice, thus a recent study evaluated changes in energy metabolism in T2DM patients treated with colesevelam. However, energy expenditure was not affected by colesevelam treatment, and no association between plasma bile acids and energy expenditure was observed. 52 More investigations will be necessary to clarify whether bile acids have an impact on energy metabolism in humans and whether bile acid sequestration can modulate this putative effect.
Conclusions
BAS have proven to be useful glucose-lowering agents in T2DM patients. First, evidence from clinical and animal studies suggests that sequestrants induce the release of incretin hormones, which may contribute to this effect. Additionally, bile acids have been reported to regulate hepatic glucose metabolism, peripheral insulin sensitivity and energy metabolism by FXR- and TGR5-dependent pathways. If and to what extent these mechanisms underlie the glucose-lowering effect of bile acid sequestrants need to be evaluated in further animal and clinical studies.
Footnotes
Acknowledgments
The authors are supported by the European Union grant HEPADIP (no. 018734) and have also received funding from Daiichi Sankyo, Inc.
Author Disclosure Statement
The authors disclose a conflict of interest with Daiichi Sankyo, Inc.