
Abstract

Introduction
Several studies have shown that GLP-1 or GLP-1 analogs limit myocardial infarct size in small nondiabetic animal models (mouse, rat, rabbit). 2 –14 Only three studies assessed the effects of GLP-1 on infarct size in pigs. One study showed that GLP-1 does not reduce myocardial infarct size 15 ; another suggested that the GLP-1 analog liraglutide does not affect myocardial infarct size 16 ; and the third reported that the GLP-1 analog exenatide reduces infarct size. 17 However, it has recently been shown that intravenous exenatide, a GLP-1 receptor agonist, started before reperfusion, increased myocardial salvage in patients who presented with ST-segment elevation myocardial infarction and were treated with primary percutaneous coronary intervention. 18
Additional studies have suggested that DPP4 inhibition may also reduce myocardial infarct size in nondiabetic animals 19,20 or obese prediabetic rats. 21 However, myocardial protection by preconditioning and postconditioning is hampered in diabetic animals. 22 More recently, we have shown that administration of exenatide 23 or MK0626, an oral DPP4 inhibitor, 24 prior to 30 min ischemia/24 h reperfusion limits myocardial infarct size in the Western diet–fed Db/Db mice (a model of type 2 diabetes mellitus).
Mechanisms of Protection
Activation of the GLP-1 receptors leads to activation of adenylyl cyclase with subsequent generation of cyclic adenosine monophosphate (cAMP), and an increase in intracellular calcium, with downstream activation of protein kinase A (PKA) and phosphorylation of cAMP response element-binding protein (CREB) (Fig. 1). 22 In addition, it has been reported that GLP-1 receptor activation leads to activation of prosurvival signaling pathways, including phosphoinositide 3-kinase (PI3K), ERK1/2, glycogen synthase kinase-3β (GSK-3β), protein kinase C, and AMP-activated protein kinase (AMPK). 22,25 All of the above-mentioned proteins are involved in protection against ischemia–reperfusion injury.
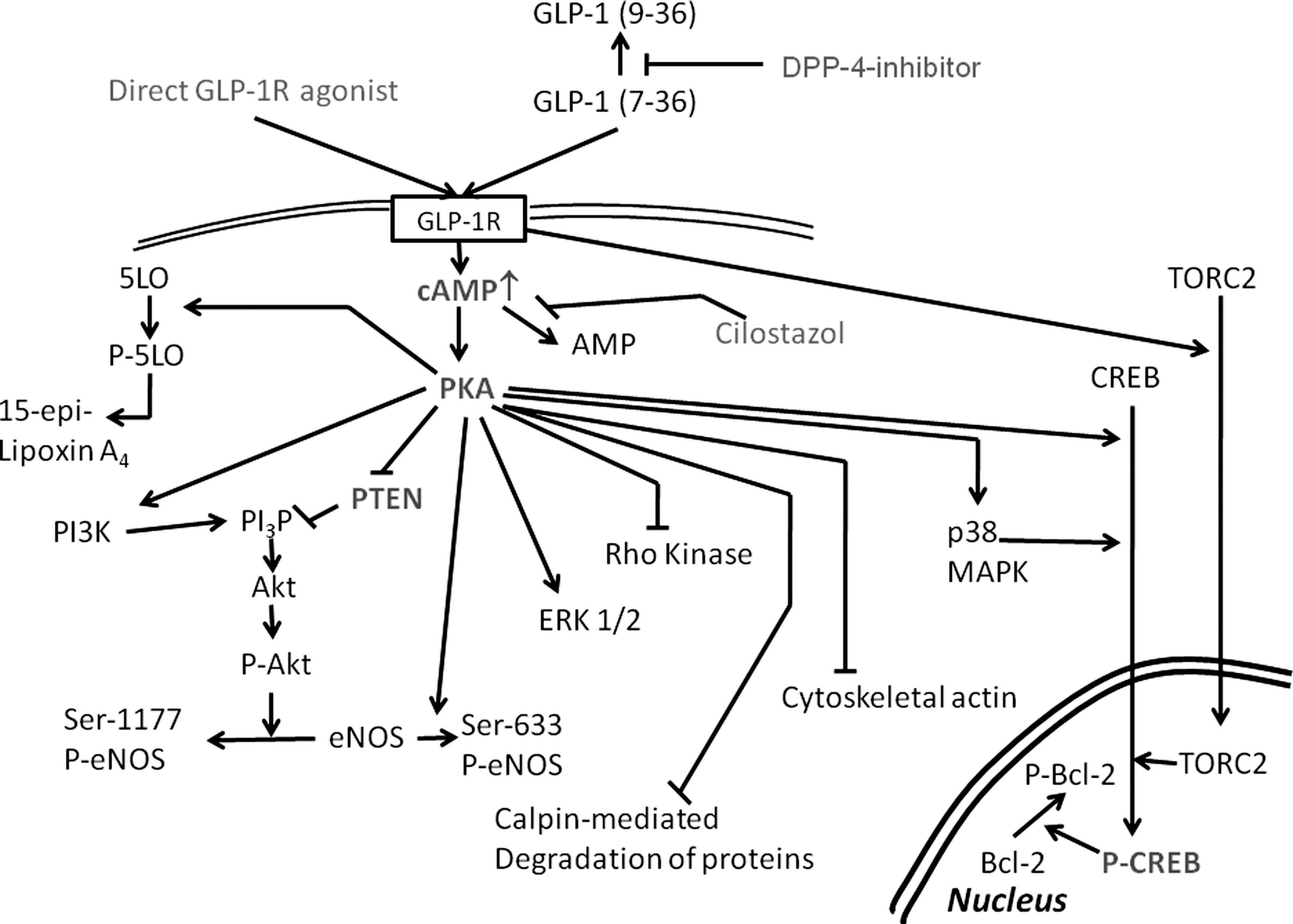
Schematic presentation of possible signaling pathways downstream of the glucagon-like peptide-1 (GLP-1) receptor activation that are involved in myocardial protection against ischemia–reperfusion injury. CREB, cyclic AMP response element binding protein; eNOS, endothelial nitric oxide synthase; GLP-1R, GLP-1 receptor; 5LO, 5-lipoxygenase; PKA, protein kinase A; PI3P, phosphoinositol 3-phosphate; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog deleted from chromosome 10.
However, it seems that GLP-1 may also have GLP-1 receptor–independent effects. 7 Although blocking the GLP-1 receptor with exendin(9-39) attenuates the infarct size–limiting effects of GLP-12,5 and the GLP-1 analog exendin-4, 6 both GLP-1 and exendin-4 improved recovery of left ventricular function after reperfusion and decreased lactate dehydrogenase (LDH) release (a measure of infarct size) in the isolated heart model of GLP-1R−/− mice. In DPP4-defficient mice, exendin-(9-39) did not completely block the infarct size-sparing effects of DPP4 deletion. 20 These findings suggest that some of the protective effects may be GLP-1 receptor–independent. In contrast, it was reported that liraglutide did not increase Akt and GSKβ phosphorylation in GLP-1R−/− mice, as it did in wild-type mice. 10 More recently, it was reported that the GLP-1 analog exendin-4 did not protect cardiomyocytes that were treated concomitantly with the GLP-1 receptor inhibitor exendin(9-39) and cardiomyocytes derived from GLP-1R−/− mice. 12 However, GLP-1(9-36) increased cAMP and protected cardiomyocytes from GLP-1R−/− mice, although exendin(9-39) blocked the protective effect. 12 These results suggest that other receptors may be involved, or that there are significant differences between GLP-1 analogs and native GLP-1 or the metabolite GLP-1(9-36).
In our opinion, cAMP-induced PKA activation has a central role in mediating the protective effects of GLP-1 receptor activation. PKA is involved in myocardial protection against ischemia–reperfusion injury. 26 It was suggested that the protective effect of ischemic preconditioning is caused by PKA-dependent attenuation of calpain-mediated degradation of structural proteins 27 or by upregulation of p38 mitogen-activated protein kinase (MAPK). 28 In nondiabetic mice, H-89, a PKA inhibitor, completely blocked the infarct size-limiting effects of 3-day pretreatment with sitagliptin. 19 In this model, H-89 did not inhibit sitagliptin-induced augmentation of Akt phosphorylation at Ser-473 and Thr-308, suggesting that PKA is downstream from Akt activation. 19 However, H-89 attenuated the sitagliptin-induced enhanced endothelial nitric oxide synthase (eNOS) phosphorylaton on Ser-1177 and Ser-633, suggesting that PKA may directly phosphorylate eNOS. 19 In vitro studies showed that inhibition of Akt by wortmannin, ERK 1/2 by U-0126, and PKA by H-89 abrogated the protective effects of sitagliptin+GLP-1, whereas cyclooxygenase-2 inhibition had no effect. 19 Recently, we have shown that H-89 completely blocked the infarct size-limiting effects of exenatide (administered 1 h before surgery) in Db/Db mice fed a Western diet. 23 Interestingly, H-89 abrogated the exenatide-induced upregulation of Akt and ERK 1/2 without affecting total Akt and ERK 1/2 levels. 23 These results suggest that with short-term GLP-1 receptor activation PKA is upstream of Akt and ERK 1/2 phosphorylation. Thus, there might be differences between nondiabetic and diabetic mice or between short-term (1 h for exenatide) and longer-term (3 days for sitagliptin) pretreatment. Alternatively, DPP4 inhibition may have additional non-GLP-1–dependent effects.
It has been suggested that PKA leads to Akt activation. Three different pathways have been described: (1) PKA activates the p85α regulatory subunit of PI3K and thus increases Akt phosphorylation 29 –31 ; (2) PKA physically interacts with Akt and phosphorylates it 32 ; (3) PKA activation leads to decreased phosphatase and the tensin homolog deleted from chromosome 10 (PTEN) expression. 23,24,33 PTEN degrades phosphatidylinositol 3-phosphate (PI3P) and thus, attenuates Akt phosphorylation. PKA also directly phosphorylates eNOS at Ser-1177 and Ser-633. 34 eNOS has an essential role in mediating protection against ischemia–reperfusion injury. 35
Because PKA activation is essential for mediating the protective effect, and PKA activity is dependent on cAMP levels, we assessed whether inhibition of cAMP degradation by a phosphodiesterase-3 inhibitor could augment the protective effect. Indeed, oral cilostazol had synergistic effects when added to DPP4 inhibitor 24 or exenatide 23 in the Western diet-fed Db/Db mouse. Cilostazol and GLP-1 receptor activation had additive effects on myocrdial cAMP levels, PKA activity, Akt, ERK1/2, and CREB phosphorylation. 23,24 . The combination of cilostazol with MK0626 had additive effects on eNOS phosphorylation at Ser-633. 24
CREB is a major nuclear transcription factor downstream of PKA that transduces cAMP activation of gene transcription. 36 CREB is activated by phosphorylation at Ser-133 by PKA. 36 CREB phosphorylation induces translocation of cytoplasmic CREB to the nucleus. 36 CREB is involved in ischemic 37,38 and pharmacologic 39,40 preconditioning. The promoter region of genes encoding cytochrome c and Bcl-xl carries a cAMP response element site, and CREB has been recognized as a positive regulator of these genes. 40 Das et al. 39 reported that adenosine receptor type 3 activation directly augments CREB Ser-133 phosphorylation followed by Bcl-2 phosphorylation. We have shown that both exenatide 23 and DPP4 inhibitors 19,24 augment CREB phosphorylation in mice hearts. Moreover, the combination of cilostazol with either DPP4 inhibitor 24 or exenatide 23 increased CREB phosphorylation more than when each drug was administered alone. Moreover, H-89 blocked this augmentation, confirming that PKA phosphorylates CREB. 19
The suppression of PTEN may have important implications concerning myocardial protection against ischemia–reperfusion injury. Myocardial levels of PTEN are increased in diabetic compared to nondiabetic rats, leading to reduced ability to phosphorylate Akt, and, hence, blunting of the protective effects of ischemic preconditioning. 41,42 However, because phosphorylated Akt is also involved in mediating insulin signaling and glucose transport, the effects of combining cilostazol and DPP4 inhibitors may have potentiating effects on the antidiabetic properties of the drugs. Indeed, we have shown that the combination of cilostazol with MK0626 (administered for 3 days) has additive effects on fasting serum glucose and triglyceride levels in the Db/Db mouse. 24
Activation of the cAMP/PKA signaling pathway was also found to augment the production of 15-epi-lipoxin-A4. 15-Epi-lipoxin A4 is an arachidonic acid–derived eicosanoid with potent antiinflammatory and inflammation resolution properties. 43,44 15-Epi-lipoxin-A4 is produced by cyclooxygenase-2 [which converts arachidonic acid to 15-hydroxyeicosatetraenoic acid (15-R-HETE)] and 5-lipoxygenase (which converts 15-R-HETE to 15-epi-lipoxin A4). 43,44 The antiinflammatory effects of 15-epi-lipoxin A4 may be beneficial for patients with type 2 diabetes and/or atherosclerosis. We have shown that phosphorylation of 5-lipoxygenase at Ser-523 by PKA is essential for preventing 5-lipoxygenase from interacting with cytosolic phospholipase A2 to generate proinflammatory leukotrienes and enhancing interaction with cyclooxygenase-2 to generate 15-epi-lipoxin A4. 44 Combining cilostazol with either a DPP4 inhibitor 24 or exenatide 23 results in higher cAMP levels and PKA activity. Myocardial levels of 15-epi-lipoxin A4 are higher when these drugs are combined as compared to administrating cilostazol, DPP4 inhibitor or exenatide alone. 23,24 It is unclear whether 15-epi-lipoxin A4 mediates (at least part of) the infarct size-limiting effects of GLP-1 receptor activation; however, a similar eicosanoid, resolving E1 (derived by cyclooxygenase-2 and 5-lipoxygenase from eicosapentaenoic acid) protects against myocardial ischemia–reperfusion injury in nondiabetic rats. 45
In conclusion, activation of the GLP-1 receptors leads to increased intracellular levels of cAMP with subsequent PKA activation. PKA has diverse downstream activities that confer protection against ischemia-reperfusion injury and may mediate antiinflammatory effects via phosphorylation of 5-lipoxygenase and production of 15-epi-lipoxin A4. The effects of GLP-1 receptor activation can be augmented by blocking cAMP degradation using phosphodiesterase-3 inhibitors.
Footnotes
Author Disclosure Statement
Yochai Birnbaum has received research grants from Amylin, Merck, Takeda, Roche, and Boehringer Ingelheim. Yumei Ye has received research grants from Amylin, Merck, Takeda, Roche, and Boehringer Ingelheim. Mandeep Bajaj has received research grants from Takeda, Amylin, Eli Lilly, Bristol-Myers Squibb, and Astra Zenica; honoraria for speaking from Takeda, Eli Lilly, Boehringer Ingelheim, and Sanofi-Aventis; and has served as a consultant to Takeda and Sanofi-Aventis.