
Abstract
Background:
Hyperketonemia is a pathological condition observed in patients with type 1 diabetes and ketosis-prone diabetes (KPD), which results in increased blood levels of acetoacetate (AA) and β-hydroxybutyrate (BHB). Frequent episodes of hyperketonemia are associated with a higher incidence of vascular disease. We examined the hypothesis that hyperketonemia activates the nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways that regulate intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells.
Methods:
Human umbilical vein endothelial cells (HUVECs) were cultured with AA (0–8 mM) or BHB (0–10 mM) for 0–24 hr. Western blotting was used to determine NF-κB activation in whole-cell lysates. ICAM-1 expression was measured using flow cytometry.
Results:
Results show a 2.4-fold increase in NF-κB activation in cells treated with 8 mM AA compared to the control. BHB had little or no effect on NF-κB activation. Pretreatment with a reactive oxygen species (ROS) inhibitor [N-acetyl-
Conclusions:
These results suggest that NF-κB and p38 MAPK mediate upregulation of ICAM-1 expression in endothelial cells exposed to elevated levels of AA, which may contribute to the development of vascular disease in diabetes.
Introduction
T
NF-κB acts as an important signal mediator in response to a wide variety of extracellular and intracellular stimuli. Activation of the NF-κB/Rel transcription family plays a central role in the inflammation associated with diabetes and other inflammatory diseases. 18,19 Exposure of cells to various inflammatory stimuli results in rapid phosphorylation, degradation of the inhibitor of NF-κB, IκBα, and rapid translocation of NF-κB into the nucleus. Target genes induced by NF-κB activation include multiple cytokine and inflammatory molecules that aid in the process of clearing inflammation. Under chronic inflammatory conditions, NF-κB gene products can exacerbate inflammation and create a negative feedback mechanism that further activates inflammatory pathways. NF-κB is one of the most important regulators of proinflammatory gene expression and a target of multiple stress signals related to pathogens or other cellular stresses such as reactive oxygen species (ROS) and oxidative stress. 20 Multiple studies implicate the role of hyperglycemia in the activation of NF-κB and other stress-activated pathways. Patients with diabetes show a positive correlation between hyperglycemia and NF-κB activation in peripheral blood leukocytes. 21 Consistent with this, Nishikawa et al. showed that normalization of mitochondrial ROS, produced by hyperglycemia, blocked activation of the NF-κB pathway. 22 In addition to hyperglycemia, NF-κB has been shown to be involved in high glucose and AA-induced tumor necrosis factor-α (TNF-α) secretion in monocytes. 17
P38 MAPK activation can lead to phosphorylation of a wide array of downstream targets, including transcription factors. The major MAPKs have all been shown to be activated by osmotic perturbations derived either from glucose or other sources, such as advanced glycated end products, the poly pathway, and oxidative stress. 23 –25 The objective of this study was to examine the hypothesis that hyperketonemia activates NF-κB signaling and MAPK signaling, both of which in turn play a role in the regulation of adhesion molecules in endothelial cells. The results demonstrate that AA, but not BHB, activates the NF-κB pathway and ICAM-1 expression in endothelial cells. Activation of the NF-κB pathway is reduced when cells are treated with an antioxidant, and AA-induced ICAM-1 expression decreases when cells are treated with antioxidants or a p38 MAPK inhibitor. These results suggest that both NF-κB and MAPK signaling are involved in mediating vascular inflammatory responses to hyperketonemia and may be mediated by oxidative stress.
Materials and Methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza Walkersville, Inc. (Walkersville, MD). Cells were cultured to confluence in EGM-2 medium and 5% CO2 in a 37°C humidified atmosphere. The culture was passaged according to standard procedures. For experiments, HUVECs were used within 24 hr after reaching confluence, between passages three and 10. The vendor certified that the medium had endotoxin concentrations <0.005 EU/mL.
Western blotting
The cells were lysed for 1 hr with vortexing in RIPA buffer containing protease inhibitors. Lysates were centrifuged for 5 min at 13,000 rpm. Supernatants were collected, and the protein levels determined using bicinchoninic acid (BCA). Lysates were suspended in sodium dodecyl sulfate (SDS) sample buffer containing 4% β-mercaptoethanol. The contents were mixed and boiled at 100°C for 5 min. The samples were loaded onto an 8% Tris-SDS acrylamide gel and run at 80 V until complete separation was reached. The proteins were transferred to a nitrocellulose membrane (0.2 μM; Bio-Rad Laboratories, Hercules, CA) followed by blocking with 1% bovine serum albumin (BSA) prepared in Tris-buffered saline containing 0.25% Tween 20 (TBS-T) for 1 hr. The blot was then incubated with a primary antibody overnight followed by washing, then a 1-hr incubation with horseradish peroxidase (HRP)-conjugated secondary antibody. Protein bands were detected using enhanced chemiluminescence (ECL) detection reagents (Perkin Elmer, Boston, MA) and exposed on blue X-ray film (Phenix Research Products, Candler, NC).
Flow cytometry
Surface analysis of cell membrane proteins was done using direct staining procedures and flow cytometry. Ice-cold reagents/solutions and the addition of sodium azide were used to prevent the modulation and internalization of surface receptors. Incubations were all done at 4°C in the dark to prevent loss of signal. The cells were harvested at the end of the experiment using trypsin. They were washed two times in phosphate-buffered saline (PBS) containing 10% fetal calf serum (FCS) and 1% sodium azide. The cells were resuspended in 300 μL of ice-cold PBS, 10% FCS, and 1% sodium azide and primary antibody conjugated to fluorescein isothiocyanate (FITC). Cells were incubated 30 min at 4°C in the dark. Cells were then washed two times by resuspending them in ice-cold PBS. They were then resuspended in 350 μL of PBS, 3% bovine serum albumin (BSA), and 1% sodium azide. Cells were directly analyzed using flow cytometry as soon as possible. Cells were analyzed by collecting 10,000 events. Results were expressed as the total mean fluorescence intensity (MFI) of 10,000 cells. Controls for matched isotype of primary antibodies were analyzed to ensure no nonspecific binding. Unstained cells, both treated and nontreated, were used as a negative control, and cells with secondary antibody only were also analyzed to ensure no nonspecific secondary binding.
Reagents
All chemicals were obtained from Sigma Chemical Co. (St. Louis, MO) unless stated otherwise. Endothelial cell growth medium was purchased from Clonetics (San Diego, CA). The inhibitors SB203580 (p38 MAPK inhibitor) and SP600125 [c-Jun N-terminal kinase (JNK) inhibitor] were purchased from InvivoGen as a lyophilized solid and prepared according to manufacturer's instructions.
The following primary antibodies were used in this study: Mouse monoclonal anti-β-actin (Abcam, Cambridge, MA); mouse monoclonal ICAM-1 FITC (Santa Cruz Biotechnology, Santa Cruz, CA); mouse monoclonal immunoglobulin G1 (IgG1) (Abcam); rabbit monoclonal phospho-p65 (NF-κB) (Cell Signaling Technology, Danvers, MA); rabbit polyclonal p65 (NF-κB) (Santa Cruz); rabbit polyclonal SAPK/JNK, p38 MAPK, phospho-SAPK/JNK (Thr 183/Tyr 185), and phospho-p38 MAPK (Thr180/Tyr 182) (Cell Signaling Technology). The secondary antibodies used were goat anti-mouse conjugated to HRP (Bio-Rad Laboratories, Hercules, CA) and goat anti-rabbit conjugated to HRP (Millipore, Temecula, CA).
Statistical analysis
Results are expressed as mean±standard error (SE). The Student t-test was used to compare the differences between treatments using Sigma plot statistical software (SPSS, Chicago, IL). A p value of less than 0.05 was considered significant.
Results
AA activates NF-κB
NF-κB is a canonical signaling molecule involved in stress signaling pathways. NF-κB activation was determined by immunoblotting for the phosphorylated p65 subunit at Ser (536) and the total p65 subunit. To determine whether hyperketonemic conditions would affect the activation status of NF-κB, endothelial cells were treated with AA and BHB for either 24 hr or in a time-dependent manner. We observed a significant (2.4-fold) increase in the phosphorylation of the p65 subunit of NF-κB when the endothelial cells were treated with 8 mM AA (Fig. 1). BHB had little or no effect on NF-κB activation. The phosphorylation of the p65 subunit was also monitored in cells treated with 8 mM AA for 0–2 hr. We observed a steady increase in the phosphorylation of p65 when cells were treated from 0 to 24 hr (data not shown). Either TNF-α or lipopolysaccharide (LPS) was used as a positive control.
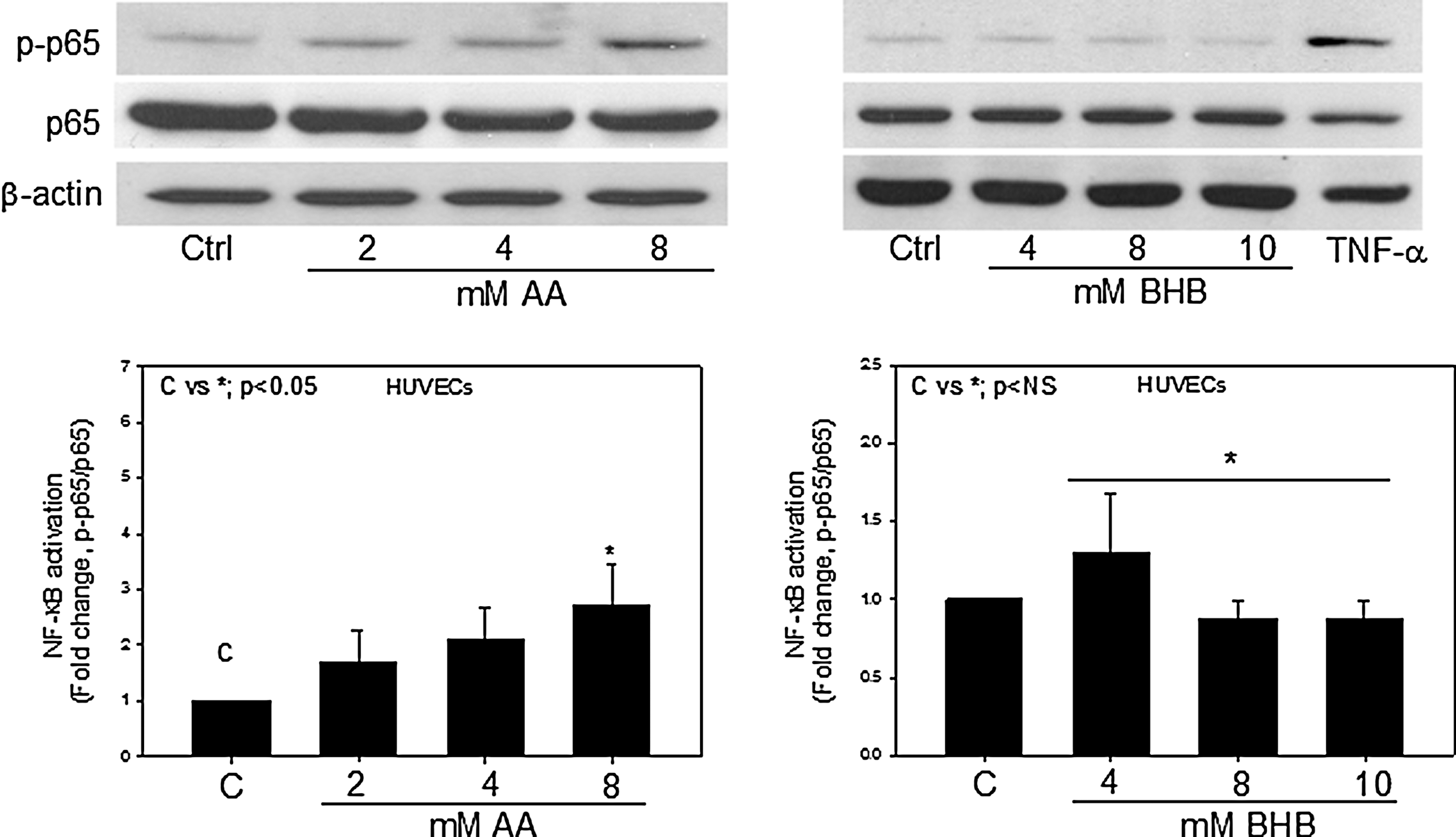
Nuclear factor-κB (NF-κB) activation in endothelial cells. Human umbilical vein endothelial cells (HUVECs) were treated with acetoacetate (AA) (0–8 mM) or β-hydroxybutyrate (BHB) (0–10 mM) for 24 hr. Western blotting for phospho-NF-κB p65 (Ser536) and total NF-κB p65 was done using total cell lysates. Tumor necrosis factor-α (TNF-α; 5 ng/mL) was used as a positive control. Values are mean±standard error (SE) (n=4).
AA and BHB induce p38 MAPK and JNK phosphorylation
To determine whether hyperketonemia has an effect on the activation of p38 MAPK or JNK, we evaluated p38 MAPK and JNK phosphorylation in endothelial cells treated with AA or BHB at different time points and compared to an untreated control. Total p38 was determined using an antibody that recognizes at least three of the four isoforms of p38; phospo-p38 MAPK (Thr180/Tyr182) was used for the active form of p38. Total JNK and phospho-SAPK/JNK (Thr183/Tyr185) were blotted to determine endogenous and active forms of JNK. We observed that both AA and BHB increased phosphorylation of JNK very early (10 min) and by 1.5-fold over the control. The activation decreased back to control levels after 30 min (Fig. 2). AA treatment increased phosphorylation of p38 MAPK at the 10-min time point and decreased back to control levels after 30 min. BHB slightly decreased p38 MAPK phosphorylation, which remained lower than the control over the 24-hr time period (Fig. 2). TNF-α was used as a positive control to activate each of the pathways.
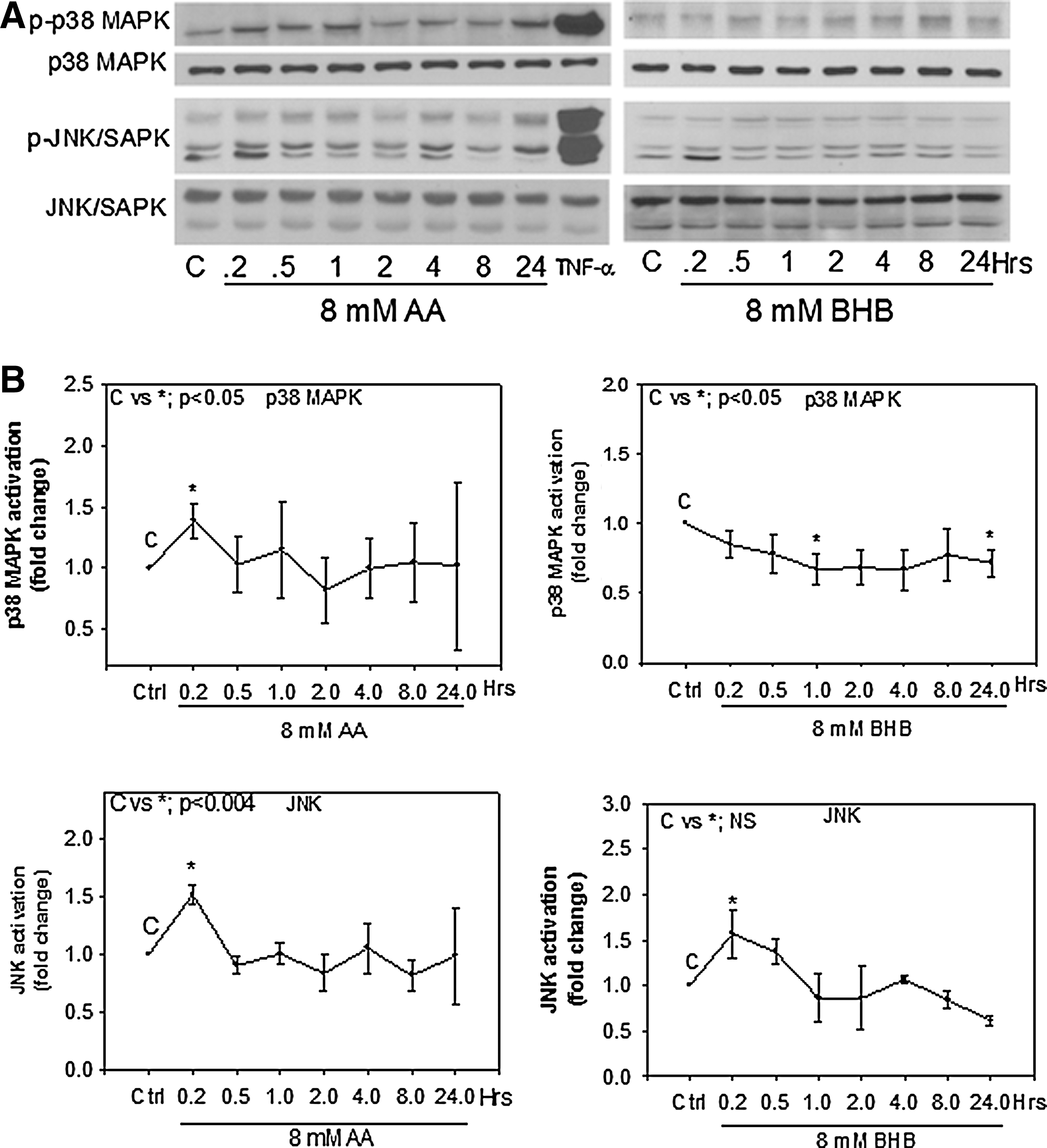
p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinases (JNK) activation in endothelial cells. Human umbilical vein endothelial cells (HUVECs) were treated with 8 mM acetoacetate (AA) or 8 mM β-hydroxybutyrate (BHB) at the indicated time points. Western blotting was done using whole-cell lysates. Antibodies used for phosphoproteins were phospho-SAPK/JNK (Thr183/Tyr185) and phospho-p38 MAPK (Thr180/Tyr182). Antibodies for total protein were SAPK/JNK and p38-MAPK. Tumor necrosis factor-α (TNF-α) was used as a positive control at 10 min. (
NAC inhibits AA activation of NF-κB
NF-κB is a redox-sensitive transcription factor and is known to be activated in oxidative stress conditions. Previous studies have indicated that AA contributes to oxidative stress conditions. 17,26 To determine whether AA-induced oxidative stress plays a role in activation of NF-κB, we pretreated cells with NAC and measured the activation of the NF-κB. Figure 3 shows that 3 mM NAC significantly reduces AA-induced NF-κB activation. These results indicate that AA induces ROS production, which may be involved in activation of the NF-κB signaling pathway.

N-acetyl-
ROS and p38 MAPK are involved in AA-induced ICAM-1 expression
Previous work has shown that AA increases ICAM-1 expression in brain endothelial cells and in HUVECs. 15,27 To determine whether ROS and MAPK signaling was involved in the regulation of AA-induced ICAM-1 expression, cells were pretreated with NAC and inhibitors to p38 MAPK (SB203580) or JNK (SP600125), and the surface expression of ICAM-1 was measured using flow cytometry. Figure 4 shows that inhibition of ROS using 3 mM NAC decreased AA-induced ICAM-1 expression to control levels. When cells were pretreated with the p38 MAPK inhibitor, ICAM-1 expression was significantly reduced to below control levels. The concentration of inhibitor used (20 μM) was similar to that used in previous studies to achieve complete inhibition of MAPK activity. 28 Pretreatment with the JNK inhibitor did not cause any significant decrease in AA-induced ICAM-1 expression (Fig. 4). These results suggest that ROS and p38 MAPK signaling are involved in stimulating the upregulation of AA-induced ICAM-1 in endothelial cells.
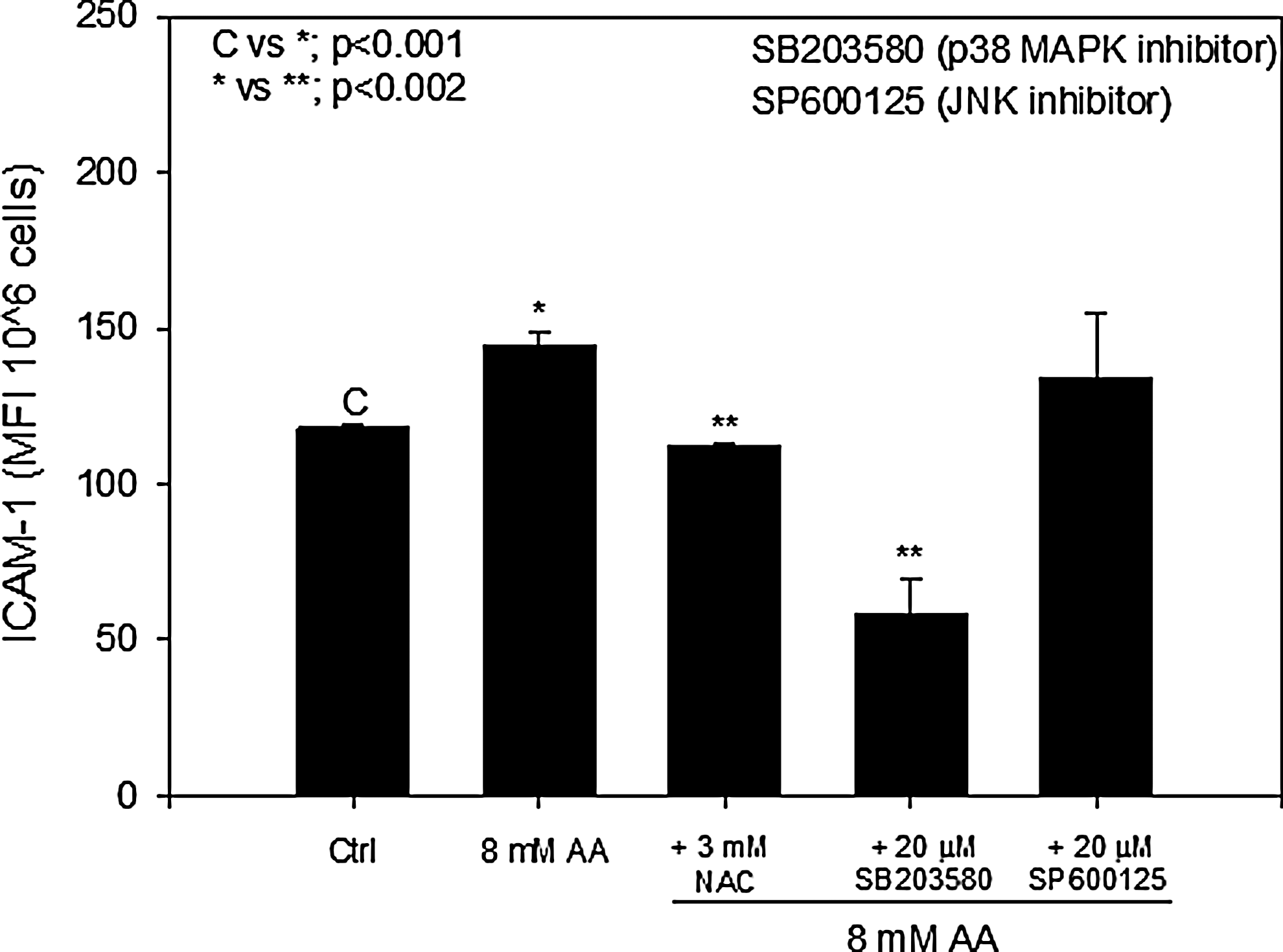
N-acetyl-
Discussion
In addition to hyperglycemia, ketosis has been shown to induce oxidative stress, which results in increases in circulating levels of various inflammatory cytokines such as TNF-α and interleukin-6 (IL-6) 17,29,30 as well as activation of the signaling molecules ERK and p38. 16 Our previous work has shown that hyperketonemia leads to adhesion molecule expression and adhesion of monocytes to endothelial cells via LFA-1/ICAM-1 interactions. 15 This study also reported that AA induced chemokine monocyte chemoattractant protein-1 (MCP-1) and IL-8 secretion in monocytes and endothelial cells, both of which play a dominant role in atherogenesis processes. 31 The literature suggests that NF-κB plays a major role in regulating the transcription of inflammatory genes whereas MAPKs regulate both the transcription and the stability of mRNA transcripts. 32,33 Recent studies in the literature also show that type 1 diabetic patients with chronic kidney disease have increased p65 activation as well as increased levels of IL-6, MCP-1, and VCAM-1. 34 No studies in the literature have examined the role of the NF-κB and MAPK pathways in response to hyperketonemia-induced inflammation. This study demonstrates that hyperketonemia can activate the stress signaling pathways NF-κB, p38 MAPK, and JNK, which may be responsible for upregulation of ICAM-1.
The results of this study show that physiological concentrations of AA can activate the NF-κB pathway in a dose-dependent manner whereas BHB has little or no effect. Activation was assayed by detection of the phosphorylated p65 subunit and compared to total protein levels of p65. BHB had a small effect on activation at a concentration of 4 mM, but the effect was not sustained at higher concentrations and showed a higher degree of variation between different experiments. These results suggest that NF-κB may be the major transcription factor involved in upregulation of other inflammatory biomarkers that have previously been shown to be influenced by hyperketonemic conditions, such as ICAM-1, TNF-α, and IL-6. 15,17,29 Because ICAM-1 is a well-documented NF-κB target gene and is upregulated in inflammatory condition, 35 –37 it is reasonable to speculate that NF-κB is involved in the regulation of AA-induced ICAM-1 expression. NF-κB is a well-defined redox-sensitive transcription factor. 38 It has been demonstrated that NF-κB activation by a wide variety of stimuli can be blocked by NAC, suggesting that the production of ROS is the dominant component leading to NF-κB activation. 39 Our results show that NAC inhibits AA-induced activation of NF-κB and ICAM-1 expression in endothelial cells. This suggests that oxidative stress may contribute to activation of NF-κB and expression of adhesion molecule ICAM-1.
MAPKs play an important role in normal signaling pathways as well as stress signaling pathways. These kinases can be activated by a number of different endogenous and exogenous cellular stress stimuli. The MAPK activation state of p38 MAPK and JNK was evaluated from 10 min to 24 hr. The results show early activation in endothelial cells that diminishes over time. MAPK activation is a very rapid process that can be diminished by the action of phosphatases just as rapidly. 40 Interestingly, BHB induced JNK phosphorylation but not p38 MAPK activation. BHB did not show any increase in NF-κB activation or ICAM-1 upregulation; therefore, the BHB-induced activation of JNK is unclear. It is possible that the ketones are directly stimulating an oxidative burst in endothelial cells, resulting in the very rapid response to MAPK activation. ERK is another MAPK family member that was not evaluated in this study; therefore, involvement of the ERK signaling pathway in hyperketonemia-induced inflammation is unknown.
Specific inhibitors to the JNK and p38 pathways were added to the endothelial cells prior to treatment with 8 mM AA. The MAPK inhibitor to p38 significantly reduced ICAM-1 expression, whereas JNK inhibition only slightly decreased expression. This suggests that AA-induced ICAM-1 expression may also be specifically regulated through p38 signaling and not through JNK signaling. ROS generation was not detected in endothelial cells at 24 hr (data not shown); however, incubation with NAC decreased AA-induced ICAM-1 expression to control levels. This suggests that oxidative stress plays a role in AA-induced ICAM-1 expression. Because pretreatment with NAC also inhibited ICAM-1 upregulation, it is unclear whether ROS is involved in activation of p38 MAPK. MAPK activation was detected early, thus it is possible that ROS production may also be very early, which would explain why we did not detect ROS at 24 hr after treatment. The ERK inhibitors U0126 and PD98059 were also used; however, there was increased expression of ICAM-1 in endothelial cells (data not shown). This could be due to toxic effects from the inhibitors alone. Further studies are needed to determine the role of ERK in hyperketonemia.
NF-κB has been implicated in every stage of diabetes and is one of the most commonly studied inflammation-causing targets. 18,19 It is a target of multiple stimuli, and constant activation can lead to a chronic inflammatory state. MAPKs are among another set of signaling molecules that have been implicated in all stages of diabetes and other inflammatory diseases. They possess a wide range of stimuli and signal responses. Both NF-κB and MAPKs represent canonical inflammatory signaling pathways, although they may serve different functions upon activation. Activation of multiple stress signaling pathways is common among many inflammatory diseases. This study demonstrates that physiological concentrations of AA can contribute to the overall inflammatory state in diabetes by increasing activation of the NF-κB and p38 MAPK pathways. Furthermore, activation of these pathways may be responsible for increased adhesion molecule expression and the increase in adhesion of monocytes to endothelial cells under hyperketonemic conditions. The results presented in this paper suggest that patients with frequent episodes of hyperketonemia would benefit from a tailored antioxidant treatment. Further evidence is needed to assess the impact these pathways have on hyperketonemia-induced adhesion. Overall, these data further support the idea that hyperketonemia contributes to the inflammatory states and the risk for cardiovascular disease in diabetes.
Footnotes
Acknowledgments
The authors are supported by grants from National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the Office of Dietary Supplements of the National Institutes of Health (NIH) (R01 DK072433) and the Malcolm Feist Endowed Chair in Diabetes. The authors thank Ms. Georgia Morgan for excellent editing of this manuscript. None of the authors has any financial interest in the publication of this manuscript, nor have they received any money from any other sources except the National Institutes of Health or LSU Health Sciences Center (LSUHSC).
Author Disclosure Statement
No competing financial interests exist.